


Contents
Scroll to:
 E. N. Chernyaeva,
K. V. Morozov,
A. D. Matsvay,
M. S. Guskova,
A. Y. Nekrasov,
I. F. Stetsenko,
A. O. Nosova,
O. G. Kurskaya,
A. M. Shestopalov,
G. A. Shipulin
E. N. Chernyaeva,
K. V. Morozov,
A. D. Matsvay,
M. S. Guskova,
A. Y. Nekrasov,
I. F. Stetsenko,
A. O. Nosova,
O. G. Kurskaya,
A. M. Shestopalov,
G. A. Shipulin https://doi.org/10.47183/mes.2024-26-3-40-50
Scroll to:
Introduction. Over the past two years, there has been an increase in measles morbidity in Russia and other countries. In order to assess the heterogeneity of clinically significant strains of Measles morbillivirus to reveal the sources of infection and transmission routes strains, a molecular genetic study thus becomes an urgent task. In this paper, a genetic identification of clinical strains of measles virus detected in 2023–2024 is compared with global variants as well as Russian strains detected in previous years.
Materials and methods. Forty measles virus genome sequences isolated from nasopharyngeal swab samples obtained in Moscow and Novosibirsk in 2023–2024 were included in the study. The data were then compared with strains collected in Russia in May 2023 and deposited at the Central Research Institute of Epidemiology, as well as strains collected in 2020 and 2021 in Moscow.
Results. The nucleotide sequences of studied Measles morbillivirus strains were categorized into different phylogenetic groups within genotype D8. For samples of genotype B3 collected in 2020 and 2023, a comparative analysis was performed to identify the region of origin. Phylogenetic analysis of Russian and foreign variants of measles virus suggests that strains currently circulating in Russia may be a variety of strains that had previously circulated in other countries and independently spread to Russia in 2023. After analyzing the most frequent nucleotide substitutions in various measles virus genes, the most variable genes were identified to provide a basis for the extension of phylogenetic analysis.
Conclusions. The proposed approach to molecular genetic testing of complete and partial genome sequences of clinical isolate of measles virus detected in 2023–2024 in Moscow and Novosibirsk made it possible to identify strain subgroups that differ in origin. The comparison of the Measles morbillivirus strains sequenced in the present research with global sequences allowed us to detect similar sequences identified both in 2023 and in previous years in various countries of the world. The analysis of epidemiologically significant strains of Measles morbillivirus shows that N gene can be used to reliably determine the main genotype; however, this approach is not sufficient for studying the transmission pathways of the virus.
Chernyaeva E.N., Morozov K.V., Matsvay A.D., Guskova M.S., Nekrasov A.Y., Stetsenko I.F., Nosova A.O., Kurskaya O.G., Shestopalov A.M., Shipulin G.A. Molecular and genetic characteristics from phylogenetic analysis of Russian and foreign variants of measles virus 2020–2024. Extreme Medicine. 2024;26(3):40-51. https://doi.org/10.47183/mes.2024-26-3-40-50
Measles is a highly infectious acute disease caused by an RNA virus belonging to the genus Morbillivirus in the family Paramyxoviridae. Despite the existence of a safe and effective vaccine, measles remains one of the leading causes of child mortality worldwide.
According to WHO, in 2022, after years of declining measles vaccination coverage, the number of measles cases increased by 18%, while the number of deaths due to measles infection increased by 43% (compared to data for 2021, when the number of deaths was 128,000) [1]. The incidence of measles in Russia in previous years ranged from 100 cases in 2010 to 4,500 cases in 2019, which were distributed in more than 60 regions of the country. Only one case was registered in 2021 and 100 cases in 2022 [2]. During the SARS-CoV-2 pandemic, more than 61 million doses of measles vaccine were delayed or missed in a number of countries around the world, including as part of routine childhood vaccination. For this reason, an increase in the vulnerable proportion of the population, and consequently the risk of localized measles outbreaks was only to be expected [3][4].
In early 2023, measles outbreaks were detected in almost all regions of the world. By the end of November 2023, more than 42,200 cases of measles virus infection had been reported in the European Union, five of which were fatal [5][6]. According to Russian Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing (Rospotrebnadzor), more than 13,000 people (8.9 per 100,000 population) contracted measles in 44 regions of Russia in 2023. Most cases (67.9%) of the total number of registered cases were detected in children aged 0 to 17 years inclusive, two of which were fatal. In 2023, 2,244 cases of measles (17.18 per 100,000 population) were registered in Moscow; in the same year, 266 cases of measles (9.5 per 100,000 population) were registered in Novosibirsk [7]. The head of the Russian Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing (Rospotrebnadzor) noted that, since all outbreaks were local and quickly controlled, no global restrictive measures were required. [8].
In addition to local outbreaks, migrant and refugee flows can pose a great threat to Russia. According to the UN, as of July 2022, about 1.8 million Ukrainian refugees had arrived in Russia [9]. According to WHO, in the European region Ukraine had the worst situation in terms of detected measles cases in recent years (more than 57,000 cases were reported in 2019) [10]. In 2023, large measles outbreaks also occurred in Russia’s neighboring countries with which there is reduced migration control, such as Azerbaijan, Kazakhstan, Uzbekistan, Tajikistan and Armenia. Some of these countries had also experienced measles outbreaks in previous years, such as Uzbekistan in 2019 [11]. Since this situation may lead to a significantly increased incidence of measles in Russia in the near future, is crucial to monitor the spread of different strains of measles virus and conduct timely phylogenetic analysis to determine the origin of each specific strain.
In order to trace and determine the origin of a measles virus strain, genotyping by sequencing is most commonly carried out on the nucleoprotein gene, which is considered to be the most variable. The sequencing of 450 nucleotides encoding 150 amino acids of the C-terminal domain of the nucleoprotein (N450) is the most common approach for genotyping measles virus strains. Over 11,000 nucleotide sequence records for this domain have been uploaded to the MeaNS2 database from 2019–2022 alone [12]. However, the N450 sequence is often insufficient to separate endemic strain lineages circulating in neighboring regions of the country. The resolution of measles molecular epidemiologic analysis can be improved by using a larger measles virus genome fragment for nucleotide sequence analysis. Although full-genome sequencing may be most effective in this case, this method has the significant disadvantage of high cost. The feasibility of extended genome fragment sequencing should be evaluated in comparison with full-genome sequencing data.
In this work, we aim to carry out a molecular genetic testing of measles virus strains detected in Russia during the period of increased morbidity in 2023–2024 in the major cities of Moscow and Novosibirsk and conduct their further phylogenetic analysis.
40 clinical specimens were collected from Novosibirsk and Moscow patients diagnosed with measles during outbreaks that occurred in 2023–2024. They were collected at the Federal Research Center for Basic and Translational Medicine (Novosibirsk) and the Center for Strategic Planning and Management of Biomedical Health Risks of the Federal Medical and Biomedical Agency (FMBA). Archival samples identified in Moscow in 2020 were also investigated. Genotyping was performed to identify the strain of measles virus for all samples included in the study. For genetic typing of measles virus, the data of comparative analysis of nucleotide sequences of the C-terminal domain of the nucleoprotein N gene (450 nt (nucleotides), 1,126–1,575 nt) are used; in order to achieve a more detailed characterization, fragments (850 nt) of the hemagglutinin H gene were additionally analyzed in some samples (n = 15). Since these genes are the most variable structural genes of the virus, they are best suited for determining the origin and pathways of individual measles virus strains. In addition, full genome sequencing was performed for three measles virus samples. The data obtained were compared with strains collected in Russia in May 2023 and deposited in the Central Research Institute of Epidemiology (see Table 1 for a complete list of measles virus samples from Russia used in this work).
For the comparative phylogenetic analysis of measles virus, both N and H gene fragments and full-genome sequencing data strains prevalent in Russia in 2020–2024, as well as global genetic variants, were used. Measles virus samples obtained from 40 patients from Moscow (n = 11) and Novosibirsk (n = 29) in 2023–2024, as well as two samples from Moscow detected in 2020 and 2021, were included in the analysis. All samples were obtained with full informed consent of the donors and in compliance with all necessary ethical standards.
Viral RNA isolated from clinical nasopharyngeal swab samples using the previously developed Ribo-prep purification kit was subjected to reverse transcription using AmpliTest Revert (AmpliTest, Russia) [13]. Fragments of the C-terminal domain of the N-gene as well as a fragment of the H-gene were amplified using LongAmp® Taq DNA polymerase (New England Biolabs, USA) and primers described in Table 2. Sanger sequencing was performed using BigDye™ Terminator v3.1 cyclic sequencing kit (Thermo Fisher Scientific, USA) and Applied Biosystems 3500 genetic analyzer (Thermo Fisher Scientific, USA). Three samples were sequenced by full-genome sequencing: total RNA was used for library preparation using NEBNext® Ultra™ II RNA Library Prep kit (NEB, USA); sequencing was performed using MiSeq platform and MiSeq Reagent Kit v2 (500 cycles) (Illumina, USA). The primers were taken from Schulz et al. and further optimized for our task [14].
As well as the full-genome sequences of six Russian measles virus samples isolated in 2023 and studied at the Central Research Institute of Epidemiology obtained from the NCBI GeneBank database [15], the gene sequences of N and H strains detected in 2019–2024 in different countries were additionally selected, along with the full-genome sequences for the same period from the open GenBank database. To permit a deeper comparison, the genome sequences of vaccine strains available in the GenBank database [15] were added to the study.
Metagenomic assembly of complete measles virus genome sequences was performed using Trimmomatic v0.39 software to remove adapter sequences and SPAdes Genome Assembler v3.15.5 to assemble the viral genome [16]. The sequences of N and H gene fragments obtained by Sanger sequencing were combined into one for further alignment and analysis.
Multiple alignment was performed using the MAFFT v7.505 package [17]. Phylogenetic analysis was performed using the FastTree program version 2.1.11 SSE3 [18] using the GTR+CAT model (a general time reversible model with a fixed rate for each site (an approximation of the CAT model)) and 100 bootstrap replications. The dolphin measles virus genome sequence (AJ608288) was used as an outgroup for cladogram generation.
Phylogenetic trees were analyzed using the FigTree v.1.4.4 program. The results of comparative analysis were visualized using the Matplotlib 3.6.2 and Seaborn 0.13.2 packages for the Python programming language.
According to the results of genotypic analysis, most of the samples (n = 44) collected in 2023–2024 turned out to belong to genotype D8 and only one to genetic variant B3 (Table 1). Phylogenetic analysis of partial sequencing of the N gene (Fig.1) showed that most of the samples from Russia formed a cluster within the D8 genotype, which also included some strains found abroad:
This phylogenetic cluster includes two subclusters, one of which was formed by samples from Novosibirsk (A), while the second cluster includes three samples from Moscow and two from Novosibirsk (B). The strains sequenced formed a phylogenetic group with samples Mea2 and Mea43 from Moscow.
Only two sequences of the N gene of measles virus samples detected in 2023 in Russia could be classified as representatives of phylogenetic group B3 — Mea48 registered in Novosibirsk and OR290098 sequenced by the Central Research Institute of Epidemiology with an unknown region of collection (Fig. 1, cluster B). These samples did not form a cluster with the gene sequences of viral strains detected in Russia in 2020 and 2021. In addition to the Russian samples, three more strains from Brazil, USA and Iran detected in 2023, as well as two strains from China and Switzerland detected in 2024, fell into phylogenetic cluster B3. Most Measles morbillivirus strains assigned to genotype B3 were detected before 2023.
According to phylogenetic analysis of N and H gene fragments, the Russian samples formed a subcluster within the D8 genotype. The results were used to clarify the phylogenetic structure of the population of the studied Russian strains. In particular, subcluster B within the D8 genetic group formed by samples Mea7, Mea8, Mea13, Mea44, and Mea45 was not confirmed (Figs. 2, 3) based on the analysis of the N gene alone.
The comparison of the results of phylogenetic analysis using only the N fragment and the sequence obtained by combining the sequences of the N and H gene fragments supports the conclusion that sequencing of the N gene is sufficient to determine the genetic group of measles virus strains; however, in order to study the virus transmission routes, an extended analysis involving additional genome regions is necessary. An additional study of the H gene allows a more precise determination of genetic similarity between measles virus samples.
Complete genome sequencing was performed for two samples of genotype D8, which belong to two phylogenetically distant and identified in different cities strains: Mea1 (Moscow, 2023) and Mea2 (Novosibirsk, 2023). Full-genome sequencing was also performed for the Mea4 sample obtained in 2020 and belonging to genotype B3. Comparative analysis of the genomes revealed 145 nucleotide differences between Mea1 and Mea2, of which 39 fall in the intergenic space and 106 substitutions in various protein-coding fragments of the genes. The highest number of these are in the L (52 substitutions), N (17 substitutions), and H (15 substitutions) genes. This result suggests that these measles virus strains may have different origins.
For phylogenetic analysis of full-genome sequences, all available genomes from the GenBank database identified in the last 5 years and belonging to groups B3, D8, as well as sequences of vaccine strains, were included in the study. Phylogenetic analysis of full genome sequences of Russian strains with sequences found in other regions of the world showed that Russian strains are closest to the samples isolated in the USA and India in 2019, as well as to those collected in Japan and New Zealand in 2023; the corresponding data are presented in Figure 4.
The differences in the formed subgroups within measles virus genotypes obtained from phylogenetic analysis of virus strains raise the question of the efficiency of analysis based on the N gene alone. To further investigate this issue, a comparative analysis of phylogenetic trees generated based on the N gene, N and H genes, along with full-genome sequences, was performed by estimating the Hamming distance between pairs of strains within and between the B3 and D8 genotypes. The results of the analysis (Fig. 5) reflect the discriminatory power of phylogenetic analysis based on different numbers of genes.
The results of the analysis clearly demonstrate the sufficient discriminatory power of the N gene fragment analysis in determining D8 and B3 genotypes, but insufficient efficiency in separating phylogenetic groups within genotypes. The simultaneous analysis of N and H genes allows strains to be separated both within and between phylogenetic groups with higher accuracy, thus increasing the statistical significance and reliability of the study. Analysis of all protein-coding fragments and full-genome sequences significantly increases the accuracy of the study of virus transmission pathways within the same phylogenetic group (Fig. 5).
To assess the variability of Measles morbillivirus genes, we performed a comparative analysis of gene sequences in the study sample consisting of full-length genomes of measles virus strains from different countries belonging to the D8 and B3 genetic groups. The results of the comparative analysis were used to determine the total number of single-nucleotide variants in the genes of proteins N, L, M, F, PVC and H. The number and proportion of synonymous and nonsynonymous nucleotide variants were estimated (Fig. 6). The highest number of nucleotide substitutions was found in the L gene, which is the largest and encodes a viral RNA polymerase protein of 2183 amino acids. The distribution profile of mutations in the M gene encoding the virus matrix protein of 335 amino acids differed most markedly from the other genes. In particular, this gene had more nonsynonymous substitutions (n = 114) than synonymous substitutions (n = 71). The PVC gene, which encodes several viral proteins (phosphoprotein (P), nonstructural protein V, and protein C), was also found to contain more nonsynonymous substitutions (n = 88) than synonymous substitutions (n = 69).
As well as demonstrating that the N gene is not the most variable, the analysis of nucleotide substitution frequencies in Measles morbillivirus genes (Fig. 6) shows that the M and H genes are also important targets for clarifying phylogenetic relationships between pathogen strains.
The results of comparative analysis of the examined measles virus samples suggest genetic similarity between Russian circulating variants and strains that have been found in other regions of the world, such as Central Europe or Asia. The published data for 2019–2022 are consistent with our results. Thus, the D8 and B3 genotypes are dominant in the global pattern of measles virus genetic diversity. However, their ratio is shown to vary in different regions of the world. In particular, the B3 genotype was dominant in the African, American, and Eastern Mediterranean regions, while the D8 genotype was dominant in the European, Northeast Asian, and Western Pacific regions [19]. The information presented on the website of the Nextstrain database [20] also indicates the dominant distribution of B3 and D8 genotypes in different regions of the world from 2020 (Fig. 7).
Most of the studied variants identified by us in 2023–2024 belong to the D8 genotype, which includes a widespread group of strains [12][19]. In total, the open GenBank database contains more than 6 thousand gene sequences or genomes of strains belonging to this group.
Among Russian strains, in addition to the D8 genotype, strains belonging to the B3 genotype were also detected. Although the B3 genotype variant was detected in only two Russian samples in 2023, both samples obtained in Moscow in 2020 and 2021 belong to this group, which is currently prevalent in Africa, Europe and North America. Among the Measles morbillivirus genomes or genome fragments obtained from patient samples in 2023 and available in public databases such as the National Center for Biotechnology Information (NCBI) database, only 60 belonged to this group; these were found predominantly in the United States and Iran. The phylogenetic analysis of this group of strains shows that the variants detected in Russia in 2020 and 2023 do not form a common cluster, which suggests a different origin. The work by Erokhov et al. [12] demonstrated in the outbreaks of 2018–2020 the presence of genetic lines and variants of measles virus that probably did not circulate in Russia, but were imported from neighboring countries. A similar conclusion was reached in the retrospective analysis of measles outbreaks in the Astrakhan Oblast carried out by Kuzmenkov et al. [21], who identified the process of migration of virus variants from neighboring countries as a key factor in the formation of new outbreaks.
Based on the above, we can assume that the majority of measles outbreaks in Russia in 2023–2024 are caused by D8 variants, which were also identified in various regions of the world during this period. This is supported by statistical data on the distribution of genotypes among measles cases for the period from 2020 to 2024 in the European region [6].
Most of the studied Measles morbillivirus nucleotide sequences associated with recent measles outbreaks in Russia can be divided into phylogenetic subgroups according to the place of origin or pathway of spread within the D8 genotype. The distance dependence between subgroups (Fig. 5) confirms that the resolution of phylogenetic analysis depends on the size of the fragment of the nucleotide sequence of the virus genome used in the analysis. The comparative analysis of phylogenetic tree structures obtained by analyzing the sequence of the N gene, along with the combination of the N and H genes, showed that the use of the N gene alone is sufficient to determine the main genotype with high reliability, but does not allow for the study of virus transmission routes. The comparison of Measles morbillivirus gene variability also showed that the N gene is not the most variable gene; thus, additional genes, such as M and H, are required to investigate transmission pathways. To achieve an optimal ratio of labor and resolution in the future, at least the three most variable genes for a selected genotype, such as N, H, and M, should be used to analyze measles virus transmission pathways.
The described approach to the molecular genetic study of complete and partial genome sequences of clinical measles virus strains detected in 2023–2024 in Moscow and Novosibirsk was used to identify subgroups differing in terms of their origin. Among the sequencing data of strains detected in 2023, we also identified two samples belonging to genotype B3, which do not form a common origin group with sequences of the same genotype detected in Moscow in 2020–2021.
The comparison of Measles morbillivirus strains sequenced by us with global sequences provides a basis for detecting both those close sequences detected both in 2023 and those identified in previous years in different countries of the world.
The study of the N gene in the analysis of epidemically significant strains of Measles morbillivirus is sufficient for determining the main genotype with high reliability, but not to study the pathways of virus transmission. For this purpose, it is recommended to use the other most variable genes, such as M and H.
Table 1. List of samples collected in different Russian cities
|
No. |
Internal sample identifier |
Year of sample taken |
City of detection |
Sequenced section of the genome |
Genotype of measles virus strain |
Data source |
|
1 |
Mea01 |
2023 |
Moscow |
WGS |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
2 |
Mea02 |
2023 |
Novosibirsk |
WGS |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
3 |
Mea04 |
2020 |
Moscow |
WGS |
B3 |
Centre for Strategic Planning of FMBA of Russia |
|
4 |
Mea05 |
2023 |
Moscow |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
5 |
Mea06 |
2023 |
Novosibirsk |
N,H |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
6 |
Mea07 |
2023 |
Novosibirsk |
N,H |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
7 |
Mea08 |
2023 |
Novosibirsk |
N,H |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
8 |
Mea10 |
2021 |
Moscow |
N |
B3 |
Centre for Strategic Planning of FMBA of Russia |
|
9 |
Mea11 |
2023 |
Мoscow region |
N,H |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
10 |
Mea12 |
2023 |
Moscow |
N,H |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
11 |
Mea13 |
2023 |
Moscow |
N,H |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
12 |
Mea14 |
2023 |
Moscow |
N,H |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
13 |
Mea15 |
2023 |
Novosibirsk |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
14 |
Mea16 |
2023 |
Novosibirsk |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
15 |
Mea17 |
2023 |
Novosibirsk |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
16 |
Mea18 |
2023 |
Novosibirsk |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
17 |
Mea20 |
2023 |
Novosibirsk |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
18 |
Mea21 |
2023 |
Novosibirsk |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
19 |
Mea22 |
2023 |
Novosibirsk |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
20 |
Mea23 |
2023 |
Novosibirsk |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
21 |
Mea24 |
2023 |
Novosibirsk |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
22 |
Mea25 |
2023 |
Novosibirsk |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
23 |
Mea26 |
2023 |
Novosibirsk |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
24 |
Mea27 |
2023 |
Novosibirsk |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
25 |
Mea28 |
2023 |
Novosibirsk |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
26 |
Mea29 |
2023 |
Novosibirsk |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
27 |
Mea30 |
2023 |
Novosibirsk |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
28 |
Mea31 |
2023 |
Novosibirsk |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
29 |
Mea32 |
2023 |
Novosibirsk |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
30 |
Mea33 |
2023 |
Novosibirsk |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
31 |
Mea34 |
2023 |
Novosibirsk |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
32 |
Mea35 |
2023 |
Novosibirsk |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
33 |
Mea36 |
2023 |
Novosibirsk |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
34 |
Mea37 |
2023 |
Novosibirsk |
N |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
35 |
Mea39 |
2023 |
Moscow |
N, H |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
36 |
Mea43 |
2023 |
Moscow |
N, H |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
37 |
Mea44 |
2023 |
Moscow |
N, H |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
38 |
Mea45 |
2023 |
Moscow |
N, H |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
39 |
Mea46 |
2023 |
Moscow |
N, H |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
40 |
Mea47 |
2024 |
Novosibirsk |
N, H |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
41 |
Mea48 |
2024 |
Novosibirsk |
N, H |
B3 |
Centre for Strategic Planning of FMBA of Russia |
|
42 |
Mea49 |
2024 |
Novosibirsk |
N, H |
D8 |
Centre for Strategic Planning of FMBA of Russia |
|
43 |
OR290097 |
2023 |
No data |
Full genome |
D8 |
Central Research Institute of Epidemiology |
|
44 |
OR290098 |
2023 |
No data |
Full genome |
B3 |
Central Research Institute of Epidemiology |
|
45 |
OR290099 |
2023 |
No data |
Full genome |
D8 |
Central Research Institute of Epidemiology |
|
46 |
OR290100 |
2023 |
No data |
Full genome |
D8 |
Central Research Institute of Epidemiology |
|
47 |
OR290101 |
2023 |
No data |
Full genome |
D8 |
Central Research Institute of Epidemiology |
|
48 |
OR290102 |
2023 |
No data |
Full genome |
D8 |
Central Research Institute of Epidemiology |
Table prepared by the authors using their own data
Table 2. List of primer sequences for the sequencing of the N and H genes
|
Sequenced gene |
Direct primer nucleotide sequence |
Nucleotide sequence of reverse primer |
|
gene N |
TGGAGCTATGCCATGGGAGT |
TAACAATGATGGAGGGTAGG |
|
gene H (region 1) |
CTGAAATTGTCTCYGGCTTC |
GACCCAGGATTGCATGTC |
|
gene H (region 2) |
GATTTCAGCAACTGYATGGT |
CTGATGTCTGRGTGACATCATG |
Table prepared by the authors using their own data
According to the results of genotypic analysis, most of the samples (n = 44) collected in 2023–2024 turned out to belong to genotype D8 and only one to genetic variant B3 (Table 1). Phylogenetic analysis of partial sequencing of the N gene (Fig.1) showed that most of the samples from Russia formed a cluster within the D8 genotype, which also included some strains found abroad:
This phylogenetic cluster includes two subclusters, one of which was formed by samples from Novosibirsk (A), while the second cluster includes three samples from Moscow and two from Novosibirsk (B). The strains sequenced formed a phylogenetic group with samples Mea2 and Mea43 from Moscow.
Only two sequences of the N gene of measles virus samples detected in 2023 in Russia could be classified as representatives of phylogenetic group B3 — Mea48 registered in Novosibirsk and OR290098 sequenced by the Central Research Institute of Epidemiology with an unknown region of collection (Fig. 1, cluster B). These samples did not form a cluster with the gene sequences of viral strains detected in Russia in 2020 and 2021. In addition to the Russian samples, three more strains from Brazil, USA and Iran detected in 2023, as well as two strains from China and Switzerland detected in 2024, fell into phylogenetic cluster B3. Most Measles morbillivirus strains assigned to genotype B3 were detected before 2023.
According to phylogenetic analysis of N and H gene fragments, the Russian samples formed a subcluster within the D8 genotype. The results were used to clarify the phylogenetic structure of the population of the studied Russian strains. In particular, subcluster B within the D8 genetic group formed by samples Mea7, Mea8, Mea13, Mea44, and Mea45 was not confirmed (Figs. 2, 3) based on the analysis of the N gene alone.
The comparison of the results of phylogenetic analysis using only the N fragment and the sequence obtained by combining the sequences of the N and H gene fragments supports the conclusion that sequencing of the N gene is sufficient to determine the genetic group of measles virus strains; however, in order to study the virus transmission routes, an extended analysis involving additional genome regions is necessary. An additional study of the H gene allows a more precise determination of genetic similarity between measles virus samples.
Complete genome sequencing was performed for two samples of genotype D8, which belong to two phylogenetically distant and identified in different cities strains: Mea1 (Moscow, 2023) and Mea2 (Novosibirsk, 2023). Full-genome sequencing was also performed for the Mea4 sample obtained in 2020 and belonging to genotype B3. Comparative analysis of the genomes revealed 145 nucleotide differences between Mea1 and Mea2, of which 39 fall in the intergenic space and 106 substitutions in various protein-coding fragments of the genes. The highest number of these are in the L (52 substitutions), N (17 substitutions), and H (15 substitutions) genes. This result suggests that these measles virus strains may have different origins.
For phylogenetic analysis of full-genome sequences, all available genomes from the GenBank database identified in the last 5 years and belonging to groups B3, D8, as well as sequences of vaccine strains, were included in the study. Phylogenetic analysis of full genome sequences of Russian strains with sequences found in other regions of the world showed that Russian strains are closest to the samples isolated in the USA and India in 2019, as well as to those collected in Japan and New Zealand in 2023; the corresponding data are presented in Figure 4.
The differences in the formed subgroups within measles virus genotypes obtained from phylogenetic analysis of virus strains raise the question of the efficiency of analysis based on the N gene alone. To further investigate this issue, a comparative analysis of phylogenetic trees generated based on the N gene, N and H genes, along with full-genome sequences, was performed by estimating the Hamming distance between pairs of strains within and between the B3 and D8 genotypes. The results of the analysis (Fig. 5) reflect the discriminatory power of phylogenetic analysis based on different numbers of genes.
The results of the analysis clearly demonstrate the sufficient discriminatory power of the N gene fragment analysis in determining D8 and B3 genotypes, but insufficient efficiency in separating phylogenetic groups within genotypes. The simultaneous analysis of N and H genes allows strains to be separated both within and between phylogenetic groups with higher accuracy, thus increasing the statistical significance and reliability of the study. Analysis of all protein-coding fragments and full-genome sequences significantly increases the accuracy of the study of virus transmission pathways within the same phylogenetic group (Fig. 5).
To assess the variability of Measles morbillivirus genes, we performed a comparative analysis of gene sequences in the study sample consisting of full-length genomes of measles virus strains from different countries belonging to the D8 and B3 genetic groups. The results of the comparative analysis were used to determine the total number of single-nucleotide variants in the genes of proteins N, L, M, F, PVC and H. The number and proportion of synonymous and nonsynonymous nucleotide variants were estimated (Fig. 6). The highest number of nucleotide substitutions was found in the L gene, which is the largest and encodes a viral RNA polymerase protein of 2183 amino acids. The distribution profile of mutations in the M gene encoding the virus matrix protein of 335 amino acids differed most markedly from the other genes. In particular, this gene had more nonsynonymous substitutions (n = 114) than synonymous substitutions (n = 71). The PVC gene, which encodes several viral proteins (phosphoprotein (P), nonstructural protein V, and protein C), was also found to contain more nonsynonymous substitutions (n = 88) than synonymous substitutions (n = 69).
As well as demonstrating that the N gene is not the most variable, the analysis of nucleotide substitution frequencies in Measles morbillivirus genes (Fig. 6) shows that the M and H genes are also important targets for clarifying phylogenetic relationships between pathogen strains.
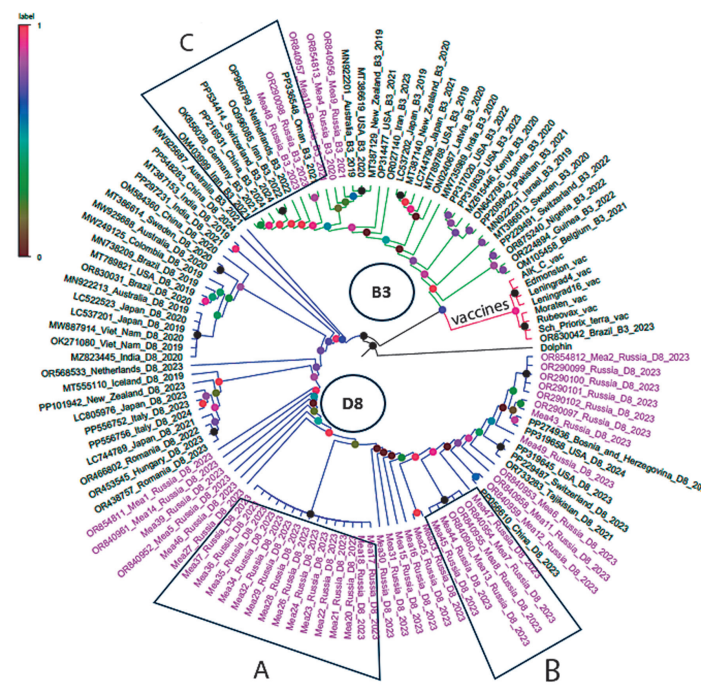
Figure prepared by the authors using their own data
Fig. 1. Phylogenetic relationships of Measles morbillivirus strains based on N-gene fragment sequencing data.
Phylogenetic group D8 is shown in blue, phylogenetic group B3 in green, and the group of vaccine strains in red. Genomes from Russia included in the analysis and sequenced in 2020, 2021 and 2023–2024 are highlighted in pink. Subcluster A consists of samples from Novosibirsk; subcluster B consists of samples from Moscow and Novosibirsk, which belong to the D8 genotype. Subcluster C of phylogenetic group B3 includes two samples — Mea48, discovered in Novosibirsk in 2023, and OR290098, sequenced by the Central Research Institute of Epidemiology with an unknown collection region.
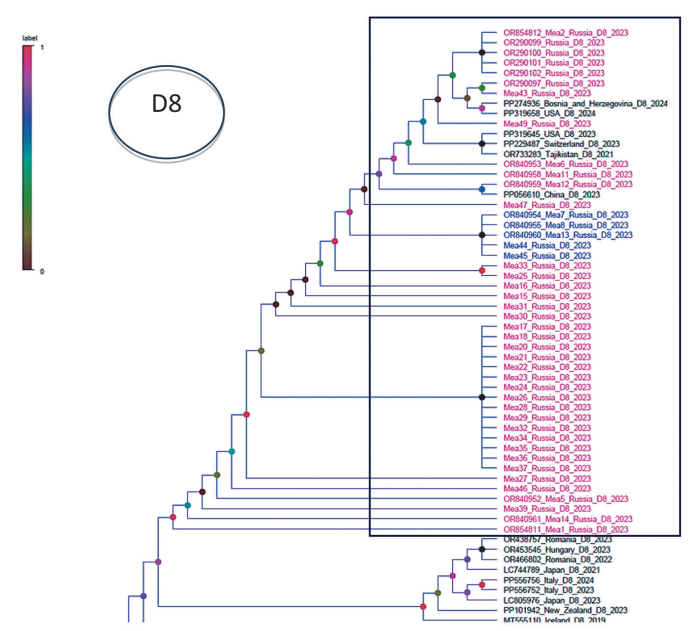
Figure prepared by the authors using their own data
Fig. 2. Position of the investigated Russian samples in cluster D8 obtained as a result of the phylogenetic analysis carried out using a fragment of the N gene sequence. The cluster formed by the studied Russian samples is highlighted with a rectangular frame; the samples are colored in blue and purple test colors. Samples Mea7, Mea8, Mea13, Mea44 and Mea45, which form subcluster B, are highlighted in blue.
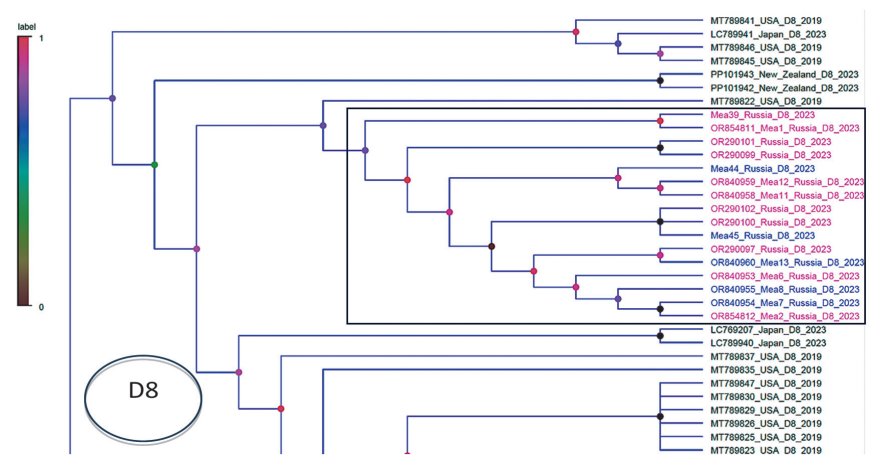
Figure prepared by the authors using their own data
Fig. 3. Position of the investigated Russian samples in cluster D8 obtained as a result of the phylogenetic analysis carried out using the combined sequence of the N and H genes. The cluster formed by the studied Russian samples is highlighted with a rectangular frame; the samples are colored in blue and purple test colors. Samples Mea7, Mea8, Mea13, Mea44 and Mea45, which form subcluster B, are highlighted in blue
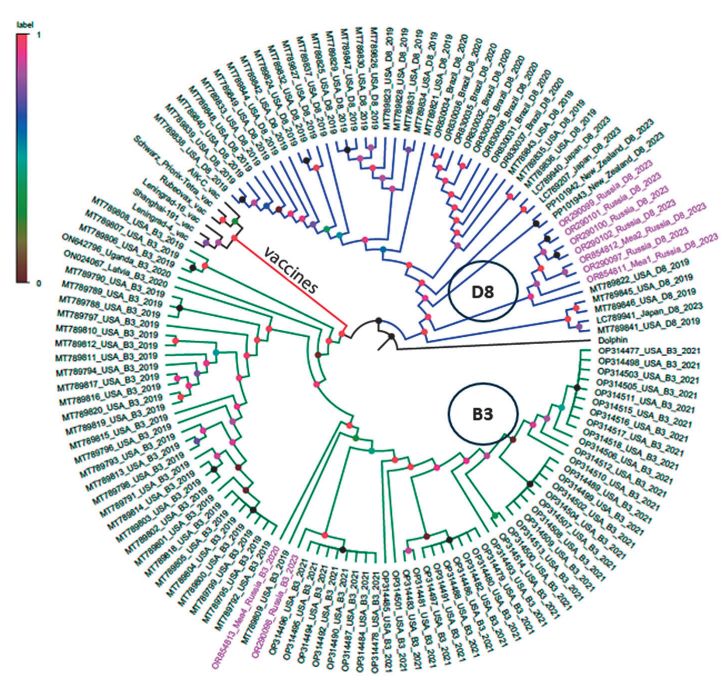
Figure prepared by the authors using their own data
Fig. 4. Phylogenetic relationships of full-genome sequences of 1 virus strains detected in Russia and other countries. The D8 genotype phylogenetic group is shown in blue, the B3 genotype group in green, and the vaccine strain group in red. Genomes from Russia included in the analysis, which were sequenced in 2020 and 2023, are highlighted in pink text
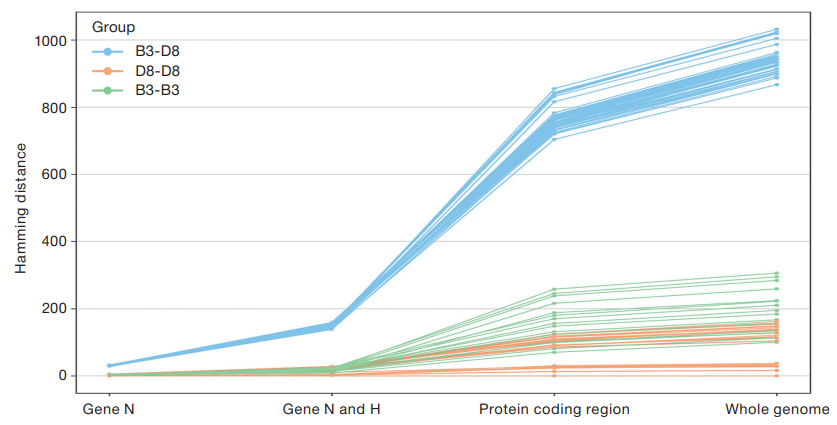
Figure prepared by the authors using their own data
Fig. 5. Pairwise Hamming distance between sequences of 1 virus strains belonging to genetic groups D8 and B3. The vertical axis shows the Hamming distance values for pairwise comparison of sequences, while the horizontal axis shows the fragments of the 1 virus genome analyzed — the N gene fragment, the union of the N and H gene fragments, and the union of all protein-coding sequences and the complete genome. The color indicates the values for the pairwise comparison of sequences within the B3 and D8 groups and between the strain groups: blue — B3-D8, green — D8-D8, orange — B3-B3
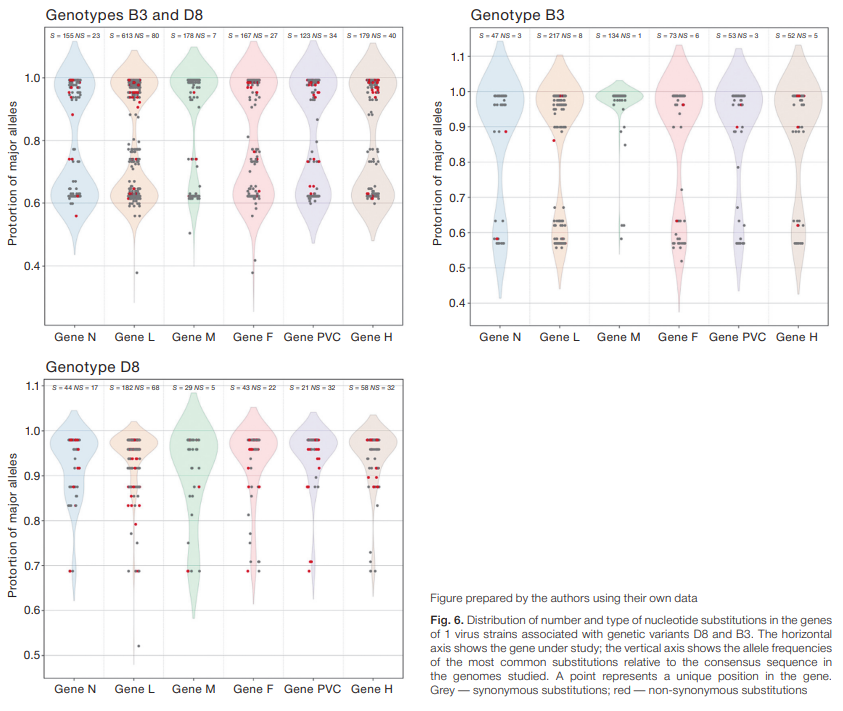
Figure prepared by the authors using their own data
Fig. 6. Distribution of number and type of nucleotide substitutions in the genes of 1 virus strains associated with genetic variants D8 and B3. The horizontal axis shows the gene under study; the vertical axis shows the allele frequencies of the most common substitutions relative to the consensus sequence in the genomes studied. A point represents a unique position in the gene. Grey — synonymous substitutions; red — non-synonymous substitutions
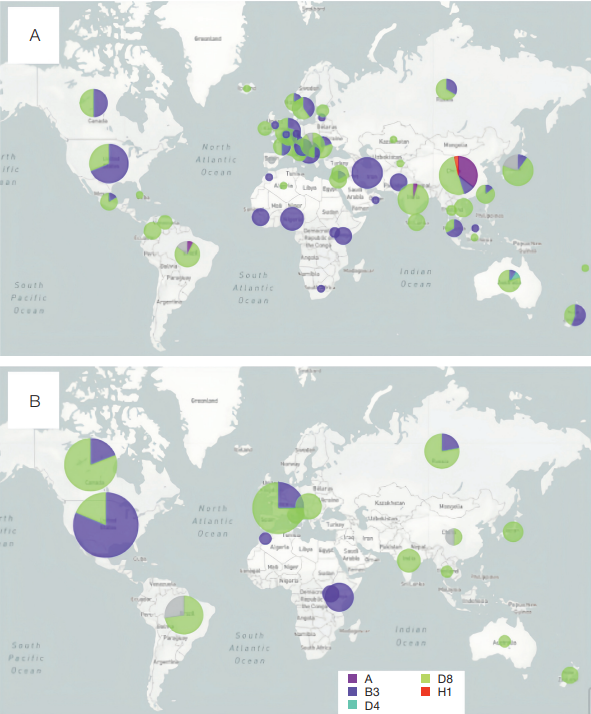
Figure prepared by the authors
Fig. 7. Global distribution of known 1 virus genotypes. Graphs based on A–N gene fragment sequencing data, B — whole genome sequencing data from January 2020 to September 2024. The figure was generated using the internet resource https://nextstrain.org/1/
The results of comparative analysis of the examined measles virus samples suggest genetic similarity between Russian circulating variants and strains that have been found in other regions of the world, such as Central Europe or Asia. The published data for 2019–2022 are consistent with our results. Thus, the D8 and B3 genotypes are dominant in the global pattern of measles virus genetic diversity. However, their ratio is shown to vary in different regions of the world. In particular, the B3 genotype was dominant in the African, American, and Eastern Mediterranean regions, while the D8 genotype was dominant in the European, Northeast Asian, and Western Pacific regions [19]. The information presented on the website of the Nextstrain database [20] also indicates the dominant distribution of B3 and D8 genotypes in different regions of the world from 2020 (Fig. 7).
Most of the studied variants identified by us in 2023–2024 belong to the D8 genotype, which includes a widespread group of strains [12][19]. In total, the open GenBank database contains more than 6 thousand gene sequences or genomes of strains belonging to this group.
Among Russian strains, in addition to the D8 genotype, strains belonging to the B3 genotype were also detected. Although the B3 genotype variant was detected in only two Russian samples in 2023, both samples obtained in Moscow in 2020 and 2021 belong to this group, which is currently prevalent in Africa, Europe and North America. Among the Measles morbillivirus genomes or genome fragments obtained from patient samples in 2023 and available in public databases such as the National Center for Biotechnology Information (NCBI) database, only 60 belonged to this group; these were found predominantly in the United States and Iran. The phylogenetic analysis of this group of strains shows that the variants detected in Russia in 2020 and 2023 do not form a common cluster, which suggests a different origin. The work by Erokhov et al. [12] demonstrated in the outbreaks of 2018–2020 the presence of genetic lines and variants of measles virus that probably did not circulate in Russia, but were imported from neighboring countries. A similar conclusion was reached in the retrospective analysis of measles outbreaks in the Astrakhan Oblast carried out by Kuzmenkov et al. [21], who identified the process of migration of virus variants from neighboring countries as a key factor in the formation of new outbreaks.
Based on the above, we can assume that the majority of measles outbreaks in Russia in 2023–2024 are caused by D8 variants, which were also identified in various regions of the world during this period. This is supported by statistical data on the distribution of genotypes among measles cases for the period from 2020 to 2024 in the European region [6].
Most of the studied Measles morbillivirus nucleotide sequences associated with recent measles outbreaks in Russia can be divided into phylogenetic subgroups according to the place of origin or pathway of spread within the D8 genotype. The distance dependence between subgroups (Fig. 5) confirms that the resolution of phylogenetic analysis depends on the size of the fragment of the nucleotide sequence of the virus genome used in the analysis. The comparative analysis of phylogenetic tree structures obtained by analyzing the sequence of the N gene, along with the combination of the N and H genes, showed that the use of the N gene alone is sufficient to determine the main genotype with high reliability, but does not allow for the study of virus transmission routes. The comparison of Measles morbillivirus gene variability also showed that the N gene is not the most variable gene; thus, additional genes, such as M and H, are required to investigate transmission pathways. To achieve an optimal ratio of labor and resolution in the future, at least the three most variable genes for a selected genotype, such as N, H, and M, should be used to analyze measles virus transmission pathways.
The described approach to the molecular genetic study of complete and partial genome sequences of clinical measles virus strains detected in 2023–2024 in Moscow and Novosibirsk was used to identify subgroups differing in terms of their origin. Among the sequencing data of strains detected in 2023, we also identified two samples belonging to genotype B3, which do not form a common origin group with sequences of the same genotype detected in Moscow in 2020–2021.
The comparison of Measles morbillivirus strains sequenced by us with global sequences provides a basis for detecting both those close sequences detected both in 2023 and those identified in previous years in different countries of the world.
The study of the N gene in the analysis of epidemically significant strains of Measles morbillivirus is sufficient for determining the main genotype with high reliability, but not to study the pathways of virus transmission. For this purpose, it is recommended to use the other most variable genes, such as M and H.
1. World Health Organization. Measles. URL: https://www.who.int/news-room/fact-sheets/detail/1#:~:text=1%20vaccination%20averted%2056%20million,the%20age%20of%205%20years (accessed: September 5, 2023).
2. Russian Statistical Yearbook, 2023. Federal State Statistics Service; 2023. URL: https://rosstat.gov.ru/storage/mediabank/Ejegodnik_2023.pdf (Accessed: July 5, 2024). (In Russ.).
3. Global Measles Outbreaks. Centers for Disease Control and Prevention. URL: https://www.cdc.gov/globalhealth/1/data/global-1-outbreaks.html (accessed: July 5, 2024).
4. Dixon MG, Ferrari M, Antoni S. Li X, Portnoy A, Lambert B, et al. Progress towards regional meats elimination — worldwide, 2000-2020. Morb Mortal Wkly Rep. 2021;70(45):1563. https://doi.org/10.15585/mmwr.mm7045a1
5. Bedford H, Elliman D. Measles rates are rising again. BMJ. 2024;384:q259. https://doi.org/10.1136/bmj.q259
6. World Health Organization. Measles. URL: https://www.who.int/europe/ru/publications/m/item/1-and-rubella-monthly-updatewho-european-region-june-2024 (accessed: July 5, 2024).
7. Measles incidence in Russia in 2023 was a 30-year record. URL: https://medvestnik.ru/content/news/Zabolevaemost-koruv-Rossii-v-2023-godu-okazalas-rekordnoi-za-30-let.html (accessed: July 5, 2024) (In Russ.).
8. RIA News. URL: https://ria.ru/20230420/kor-1866513301.html (accessed: July 5, 2024).
9. Refugees from Ukraine recorded by country. URL: https://data.unhcr.org/en/situations/ukraine (accessed: July 5, 2024).
10. The World Health Organization. 1 — number of reported cases. URL: www.who.int/data/gho/data/indicators/indicator-details/ GHO/1--number-of-reported-cases (accessed: July 15, 2023).
11. Bryantseva E, Matnazarova G, Tursunov D, Saidkasimova N. Epidemiological features of 1 infection during an outbreak in Tashkent city. EBWFF 2023 — International Scientific Conference Ecological and Biological Well-Being of Flora and Fauna (Part 1). 2023;V.420. https://doi.org/10.1051/e3sconf/202342005014
12. Erokhov DV, Zherdeva PE, Rubalskaya TS, Frolov RA, Tikhonova NT. Global genetic diversity of measles, rubella, and mumps viruses in 2019–2022. Modern problems of epidemiology, microbiology and hygiene.2022;108–11 (In Russ.). EDN: PBRIWH
13. Nosova AO, Bogoslovskaya EV, Shipulin GA. Modern approaches and prospects for the development of laboratory diagnostics of measles. Clinical microbiology and antimicrobial chemotherapy.2023;25(1). https://doi.org/10.36488/cmac.2023.1.4-12
14. Schulz H, Hiebert J, Frost J, McLachlan E, Severini A. Optimisation of methodology for whole genome sequencing of 1 Virus directly from patient specimens. Journal of Virological Methods. 2022;299:114348. https://doi.org/10.1016/j.jviromet.2021.114348
15. Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, et al. Nucleic Acids Res. 2013;41(Database issue):D36–42. https://doi.org/10.1093/nar/gks1195
16. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. Journal of Computational Biology. 2012;19(5):455–77. https://doi.org/10.1089/cmb.2012.0021
17. Katoh K, Standley DM. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Molecular Biology and Evolution. 2013;30(4):772–80. https://doi.org/10.1093/molbev/mst010
18. Price MN, Dehal PS, Arkin AP. FastTree 2 — Approximately Maximum-Likelihood Trees for Large Alignments. Plos one. 2010;5(3):e9490. https://doi.org/10.1371/journal.pone.0009490
19. Chekhlyaeva TS, Erokhov DV, Zherdeva PE, Tikhonova NT. Measles virus genotypes in the Russian Federation and their diversity in 2018–2020. Epidemiologic surveillance of current infections: new threats and challenges. 2021;327–9 (In Russ.). https://doi.org/10.21145/978-5-6046124-2-2_2021
20. Nextstrain. Real-time tracking of pathogen evolution [cited 2024 Aug. 12]; URL: https://nextstrain.org/1/ (accessed: July 5, 2024).
21. Kuzmenkov MV, Spirenkova AE, Akhmerova RR, Rvachev VS. Current epidemiologic features of measles on the territory of the Astrakhan region in 2013–2023. International Research Journal. 2024;3(141) (In Russ.).
Moscow
Moscow
Moscow
Moscow
Moscow
Moscow
Moscow
Novosibirsk
Novosibirsk
Moscow
Chernyaeva E.N., Morozov K.V., Matsvay A.D., Guskova M.S., Nekrasov A.Y., Stetsenko I.F., Nosova A.O., Kurskaya O.G., Shestopalov A.M., Shipulin G.A. Molecular and genetic characteristics from phylogenetic analysis of Russian and foreign variants of measles virus 2020–2024. Extreme Medicine. 2024;26(3):40-51. https://doi.org/10.47183/mes.2024-26-3-40-50
10 bld. 1 Pogodinskaya Str., Moscow, Russia 119121
tel.: +7 (495) 540-61-71, ext.: 4190, 4191, 4192
E-mail: Extrememedicine@cspfmba.ru
Processing of personal data











