


Содержание
Перейти к:
 А. Н. Кириенко,
Е. В. Мотыко,
Е. В. Ефремова,
Д. В. Кустова,
Т. Н. Герт,
И. В. Леппянен,
В. А. Шуваев,
И. С. Мартынкевич
А. Н. Кириенко,
Е. В. Мотыко,
Е. В. Ефремова,
Д. В. Кустова,
Т. Н. Герт,
И. В. Леппянен,
В. А. Шуваев,
И. С. Мартынкевич https://doi.org/10.47183/mes.2024-241
Перейти к:
Введение. Определение драйверных мутаций в генах JAK2, CALR и MPL является «золотым стандартом» в молекулярной диагностике пациентов с Ph-МПН. Однако геномный ландшафт таких пациентов гетерогенен, и стандартные молекулярно-генетические методы не позволяют выявить большинство соматических мутаций и тем самым не дают полного представления об особенностях течения и прогнозе Ph-МПН, а также не позволяют подтвердить клональность заболевания у больных с тройным негативным статусом. Метод секвенирования следующего поколения (NGS) дает возможность одновременно провести анализ обширной панели генов и выявить как патогенные, так и драйверные мутации.
Цель. Оценка возможности использования NGS в изучении мутационного статуса пациентов с Ph-негативными МПН и анализ влияния выявленных патогенных мутаций на выживаемость пациентов.
Материалы и методы. В исследование были включены 83 пациента с диагнозами «истинная полицитемия», «эссенциальная тромбоцитемия» и «первичный миелофиброз» в возрасте от 19 до 85 лет (медиана начала заболевания — 51 год). У всех пациентов секвенирование выполнялось с использованием миелоидной панели из 118 генов со средней глубиной прочтения 1000х на приборе MiSeq (Illumina, США). Клиническая значимость мутаций устанавливалась по базам данных COSMIC и Franklin. Для анализа выживаемости использовали метод Каплана — Мейера с оценкой статистической значимости с помощью теста Кокса — Мантела с использованием программы GraphPad Prism 8.
Результаты. Патогенные мутации в 23 генах были выявлены у 39 (46%) пациентов из общего числа больных. Наиболее часто, у 25% пациентов, мутации детектировали в гене ASXL1; они снижали бессобытийную выживаемость на 50,3% (Me = 7,83 года против 15,75 года). Выявленные патогенные мутации в других генах сочетанно с мутациями в драйверных генах также ухудшали показатели бессобытийной выживаемости по сравнению с показателями пациентов, имевших изолированные драйверные мутации. Две и более патогенные мутации значимо снижали бессобытийную выживаемость по сравнению с пациентами с одной патогенной мутацией. Методом NGS также удалось выявить патогенные мутации у 8 из 10 исследуемых пациентов с тринегативным статусом и таким образом подтвердить клональность заболевания.
Выводы. Метод секвенирования следующего поколения (NGS) с использованием панели из 118 генов является эффективным инструментом в выявлении прогностически значимых мутаций, важных для подбора наиболее эффективной персонализированной терапии, позволяющей достигать гематологического ответа.
Кириенко А.Н., Мотыко Е.В., Ефремова Е.В., Кустова Д.В., Герт Т.Н., Леппянен И.В., Шуваев В.А., Мартынкевич И.С. Изучение мутационного профиля больных Ph-негативными миелопролиферативными новообразованиями методом NGS. Экстремальная биомедицина. 2025;27(1):80-87. https://doi.org/10.47183/mes.2024-241
Kirienko A.N., Motyko E.V., Efremova E.V., Kustova D.V., Gert T.N., Leppyanen I.V., Shuvaev V.A., Martynkevich I.S. NGS analysis of the mutational profile of patients with Ph-negative myeloproliferative neoplasms. Extreme Medicine. 2025;27(1):80-87. https://doi.org/10.47183/mes.2024-241
Миелопролиферативные новообразования BCR::ABL1-негативные (Ph-МПН) представляют собой клональные гематологические злокачественные новообразования, которые характеризуются избыточным выходом зрелых миелоидных клеток в кровь и возникают из мутированной гемопоэтической стволовой клетки [1–3]. К классическим Ph-МПН относят истинную полицитемию (ИП), эссенциальную тромбоцитемию (ЭТ) и первичный миелофиброз (ПМФ) [4].
Открытия молекулярной генетики последних десятилетий позволили выявить общий «пусковой механизм» патогенеза этих заболеваний, а именно: постоянная активация янус-киназного (JAK-STAT) сигнального пути в клетке, посредством которого передается информация от внешних химических сигналов к ядру, приводит к транскрипции ДНК и экспрессии генов, участвующих в иммуногенезе, пролиферации, дифференцировке, апоптозе и онкогенезе [5]. Одной их причин постоянной активации JAK-STAT сигнального пути являются мутации в генах JAK2, CALR и MPL, получивших название «драйверных». Выявление мутаций в данных генах стало неотъемлемой частью современного диагностического алгоритма для пациентов с МПН, который включен в текущие клинические рекомендации. Расшифровка патогенетических механизмов развития Ph-МПН способствовала разработке и внедрению в клиническую практику таргетной терапии ингибиторов янус-киназ, блокирующих внутриклеточную сигнальную систему JAK-STAT [5].
В то же время исследования последних лет показали гетерогенность геномного ландшафта Ph-негативных МПН. Мутации выявляли в генах, отвечающих за различные функции внутри клетки: эпигенетическая регуляция метилирования ДНК (TET2, DNMT3A и IDH1/2), модификация гистонов/хроматина (ASXL1, EZH2, SUZ12), сплайсинг РНК (SRSF2, SF3B1, U2AF1), передача сигнала (SH2B3, LNK, CBL, RAS, NF1), факторы транскрипции (TP53 и RUNX1) и др. [6–9]. Выявление данных мутаций имеет диагностическое и прогностическое значение, позволяя оценить риск прогрессирования заболевания, выбрать наиболее эффективную тактику лечения и принять решение о необходимости трансплантации гемопоэтических стволовых клеток. Таким образом, для диагностики BCR::ABL1-негативных МПН у пациентов все большую актуальность приобретает метод секвенирования следующего поколения (next generation sequencing — NGS), позволяющий одновременно определять мутационный статус большого числа генов.
Цель исследования — оценка возможности использования метода NGS в изучении мутационного статуса пациентов с Ph-негативными МПН и анализ влияния выявленных патогенных мутаций на выживаемость пациентов.
В исследование были включены 83 пациента (30 мужчин и 53 женщины) в возрасте от 19 до 85 лет (медиана начала заболевания — 51 год), находящихся на лечении в клиниках Санкт-Петербурга и Москвы. Диагноз «Ph-негативное МПН» согласно критериям ВОЗ ранее установлен у всех больных: из них наблюдали 47 пациентов с диагнозом ПМФ, 15 больных — с диагнозом ИП, 21 человек — с диагнозом ЭТ (табл. 1).
Таблица 1. Сводные данные когорты пациентов
Основные характеристики | Количество пациентов, n |
Пол: | |
мужской | 30 |
женский | 53 |
Возраст (Me), лет | 19–85 (51) |
Диагноз: | |
Первичный миелофиброз (ПМФ) | 47 |
Истинная полицитемия (ИП) | 15 |
Эссенциальная тромбоцитемия (ЭТ) | 21 |
Мутации в драйверных генах: | |
JAK2 | 54 |
CALR | 16 |
MPL | 3 |
Тринегативный статус | 10 |
Фазовый переход / Лейкемическая трансформация: | |
ПМФ в острый миелоидный лейкоз | 7 |
ЭТ во вторичный миелофиброз | 3 |
ИП во вторичный миелофиброз | 4 |
Таблица подготовлена авторами по собственным данным
Все пациенты были обследованы ранее на наличие мутаций в драйверных генах: у 54 (65%) человек обнаруживалась мутация в гене JAK2 (V617F), у 16 (19%) — в CALR, у 3 (4%) — в MPL. Тем не менее данные гены были включены в NGS-панель исследуемых генов и использовались в качестве внутреннего положительного контроля. Группа пациентов с ЭТ и ПМФ, не имеющих мутации ни в одном из «драйверных» генов (так называемые пациенты с тринегативным статусом), составила 10 (14,7%) пациентов от общего числа выборки.
За время наблюдения у 14 пациентов был выявлен фазовый переход или лейкемическая трансформация: у 7 пациентов с диагнозом ПМФ наблюдалась трансформация в ОМЛ; у 3 пациентов с ЭТ и 4 с ИП переход во вторичный миелофиброз (табл. 1). Выделение ДНК из образцов периферической крови проводили с использованием набора QIAamp RNA Blood Mini Kit (Qiagen, Нидерланды).
Мутации в гене JAK2 определяли с помощью набора реагентов для обнаружения мутации V617F G/T гена JAK2 (Janus kinase 2) («Синтол», Российская Федерация). Мутации в генах CALR и MPL определяли методом секвенирования по Сэнгеру на генетическом анализаторе НАНОФОР-05 («Синтол», Российская Федерация). Для наработки фрагментов ДНК использовали соответствующие праймеры: MPL-F 5’-TAGCCTGGATCTCCTTGGTG-3’, MPL-R 5’-AGAGGTGACGTGCAGGAAGT-3’; CALR-F 5’-TGAGGTGTGTGCTCTGCCT-3’, CALR-R 5’-AGAGACATTATTTGGCGCGG-3.
У всех пациентов секвенирование выполнялось с использованием таргетной экзонной панели из 118 генов со средней глубиной прочтения 1000х на приборе MiSeq (Illumina, США). Самостоятельно разработанная панель включала в себя ключевые гены, задействованные в миелоидных неоплазиях [6–9]. Для секвенирования на Illumina использовали библиотеки, приготовленные из 200 нг геномной ДНК, расщепленной до фрагментов 300 п.о., с использованием фокусированного ультразвукового аппарата Covaris S2.
Фрагментированную ДНК трансформировали в ДНК-библиотеки с помощью набора KAPA Hyper Prep Kit (Roche, Швейцария). Обогащение ДНК-библиотек проводили с помощью набора Hyper Cap Target Enrichment и набора KAPA Hyper Exome Probes (Roche, Швейцария) согласно протоколу производителя. Для приготовления библиотек ДНК использовали модуль MGIEasy Circularization Module V2.0 (MGI, Китай). Количественный анализ библиотеки проводили на флуориметре Quantus с набором QuantiFluor® dsDNA System (Promega, США).
В качестве средства просмотра анализов секвенирования использовали программное обеспечение Illumina Sequence Analysis Viewer. Качество исходных данных NGS оценивалось с помощью программного обеспечения FastQC в Illumina BaseSpace Sequence Hub. Данные секвенирования были проанализированы с использованием комбинации двух приложений для выравнивания последовательностей и вызова вариантов, также применяемых в Illumina Bas eSpace Sequence Hub: приложения DNA Amplicon и приложения Pindel, с пределом обнаружения частоты аллелей 3% (VAF).
Клиническая значимость мутаций устанавливалась по базам данных COSMIC, ClinVar и Franklin согласно критериям ACMG/AMP. Для аннотации функции генов была использована база KEGG.
Для анализа выживаемости применяли метод Каплана — Мейера с оценкой статистической значимости с помощью теста Кокса — Мантела. Статистический анализ выполнялся с помощью GraphPad Prism 8 (GraphPad Software, La Jolla, CA, США).
По крайней мере одна из драйверных мутаций в генах JAK2, CALR и MPL была выявлена у 72 (87%) пациентов из общего числа больных с диагнозом «Ph-негативное МПН», принимавших участие в исследовании, что соответствует данным, полученным иными молекулярными методами.
В ходе исследования у 27 (57,5%) пациентов с диагнозом ПМФ обнаруживалась мутация в гене JAK2 (V617F), мутация в гене CALR — у 12 (25,5%), у 2 (4,2%) пациентов — мутация в гене MPL, у 6 (12,8%) пациентов не были выявлены мутации в драйверных генах (так называемый «тринегативный» статус). У всех 15 пациентов с диагнозом ИП определялась мутация в гене JAK2 (V617F). В гене JAK2 определялась только мутация в 14-м экзоне (V617F), мутации в 12-м экзоне выявлены не были. В гене MPL мутации обнаруживались только в положении W515. В гене CALR были выявлены мутации двух основных типов: делеция 52 нуклеотидов и инсерция 5 нуклеотидов.
При проведении исследования у 12 (57%) из 21 пациента с диагнозом ЭТ выявлялась мутация в гене JAK2 (V617F), мутацию в гене CALR регистрировали у 4 (19%) больных, у 1 (5%) пациента была определена мутация в гене MPL, в то же время 4 (19%) пациента имели тринегативный статус.
При NGS исследовании 118 генов мутации были выявлены у 39 (46%) из 83 пациентов в 23 генах, представленных на рисунке 1. При этом одну патогенную мутацию имели 19 (49%) из 39 наблюдаемых пациентов, 2 мутации регистрировали у 7 (18%) больных, 3 и более мутаций отмечены у 13 (33%) пациентов с Ph-негативными МПН. В среднем у всей когорты выявлялась 1 патогенная мутация.
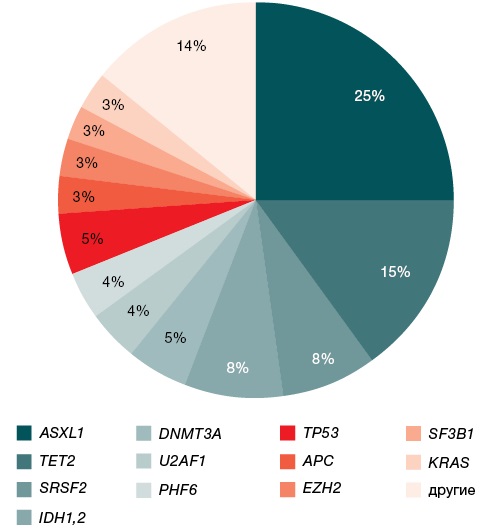
Рисунок подготовлен авторами
Рис. 1. Мутационный профиль 83 пациентов с Ph-негативными миелопролиферативными новообразованиями
В анализируемой выборке пациентов патогенные мутации выявлялись чаще других в генах ASXL1 и TET2: у 25 и 15% пациентов соответственно. Мутации в генах SRSF2 и IDH1/2 обнаруживались у 8% пациентов, DNMT3A — у 6%, U2AF1, PHF6, TP53 — у 4% каждая, APC, EZH2, SF3B1, KRAS — у 3% каждая. Мутации в генах ATRX, CBL, DDX3X, EP300, GATA2, RUNX1, SETBP1, SUZ12, ZRSR2 обнаруживались всего у 1% пациентов каждая (рис. 1).
Среди пациентов с тринегативным статусом были выявлены патогенные мутации у 8 (80%) из 10 пациентов в 7 различных генах. При этом у 4 из них была обнаружена лишь одна мутация, у 2 пациентов две мутации, у 2 пациентов три патогенных мутации одновременно. В двух генах сочетанно SRSF2 и ASXL1 мутации были выявлены у 4 пациентов с тринегативным статусом, у 2 пациентов детектировали мутацию в гене IDH1, у 1 больного — в генах RUNX1, TET2, NF1 и HRAS.
В ходе исследования были обнаружены патогенные мутации в 23 генах, которые дополнительно анализировали с помощью базы данных KEGG, позволяющей предсказать функции данных генов. Большинство генов (SRSF2, U2AF1, SF3B1, PHF6, DDX3X, ZRSR2) участвуют в сплайсинге РНК и метилировании ДНК (DNMT3A, IDH1, IDH2, TET2, SUZ12). Меньшее количество генов отвечают за модификацию хроматина (гистонов), репликацию ДНК и передачу сигналов внутри клетки; три гена выполняют роль транскрипционных факторов (табл. 2). Малый размер выборки пациентов, включенных в наше исследование, не позволил оценить влияние каждой функциональной группы на прогноз течения заболевания или эффективность терапии лечения. Тем не менее анализ с помощью базы данных KEGG способствовал пониманию молекулярных механизмов, лежащих в основе Ph-негативных миелопролиферативных новообразований.
Таблица 2. Функции генов, выявленных в ходе NGS исследования
Функции генов | Названия генов |
Модификация хроматина (гистонов) | ASXL1, ATRX, EZH2, EP300 |
Сплайсинг РНК | SRSF2, U2AF1, SF3B1, PHF6, DDX3X, ZRSR2 |
Метилирование ДНК | DNMT3A, IDH1, IDH2, TET2, SUZ12 |
Транскрипционные факторы | RUNX1, TP53, GATA2 |
Репликация ДНК | SETBP1, APC |
Передача сигнала | KRAS, CBL |
Таблица составлена авторами, аннотация функции генов проведена с помощью базы данных KEGG
В ходе исследования у 14 пациентов с фазовым переходом во вторичный миелофиброз или с лейкемической трансформацией обнаруживали в среднем 2 патогенные мутации. При этом мутации в гене ASXL1 были выявлены у 9 (64%) больных, у 3 (22%) пациентов обнаруживались мутации в генах IDH1,2, DNMT3A, TET2, у 2 (14%) — мутации детектировались в генах SRSF2, SFR3B1, TP53, у 1 (7%) пациента выявлена мутация в генах KRAS, SUZ12, PHF6.
В исследовании было показано значимое влияние мутационного профиля генов на показатели бессобытийной выживаемости пациентов. Так, мутации в гене ASXL1 статистически значимо ассоциировались (p = 0,0011) со снижением бессобытийной выживаемости (медиана — 11,8 и 15,75 года) (рис. 2) в исследуемой когорте пациентов.
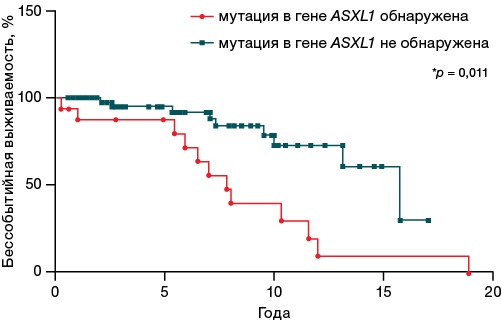
Рисунок подготовлен авторами
Рис. 2. Влияние мутаций в гене ASXL1 на бессобытийную выживаемость пациентов с Ph-негативными миелопролиферативными новообразованиями (за событие принимали фазовый переход, лейкемическую трансформацию или смерть)
При анализе данных, представленных на рисунке 3, установлено, что сочетание драйверной и любой из патогенных мутаций (группа драйверная+, патогенная+) статистически значимо связано со снижением бессобытийной выживаемости по сравнению с группой пациентов, у которых выявлены исключительно драйверные мутации (группа драйверная+, патогенная–), (медиана выживаемости — 10,0 и 15,8 года). Наихудший прогноз был показан для пациентов с тринегативным статусом и наличием хотя бы одной патогенной мутации (группа драйверная–, патогенная+), (медиана выживаемости — 6,9 года). Для группы пациентов, у которых не выявляли ни патогенные, ни драйверные мутации (драйверная–, патогенная–), медиана достигнута не была.
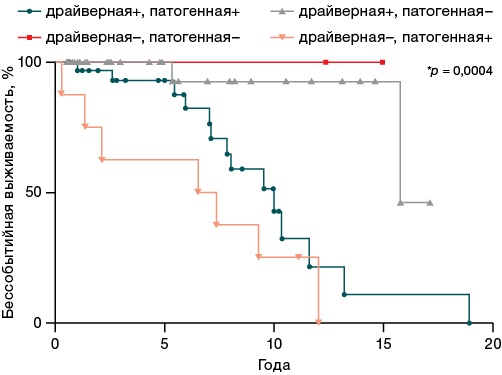
Рисунок подготовлен авторами
Рис. 3. Влияние патогенных мутаций на бессобытийную выживаемость пациентов с Ph-негативными миелопролиферативными новообразованиями
На рисунке 4 показано, что наличие двух и более мутаций также ассоциировалось с достоверным снижением бессобытийной выживаемости (p < 0,0001) (медиана выживаемости — 7 и 13,17 года) по сравнению с пациентами с меньшим количеством патогенных мутаций. Анализ данных NGS показал, что большее количество патогенных мутаций связано с ухудшением прогноза выживаемости.
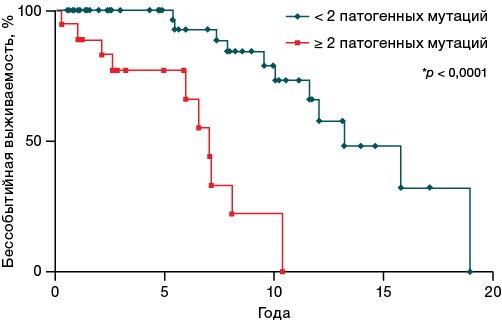
Рисунок подготовлен авторами
Рис. 4. Влияние количества патогенных мутаций на бессобытийную выживаемость пациентов с Ph-негативными миелопролиферативными новообразованиями
В основе патогенеза Ph-негативных миелопролиферативных новообразований лежат мутации в драйверных генах — JAK2, CALR и MPL. Выявление мутаций в данных генах на сегодняшний момент — основа молекулярной диагностики пациентов с подозрением на Ph-негативное МПН. Между тем исследования последних лет показывают, что генетический ландшафт классических Ph-негативных МПН не ограничивается только мутациями в драйверных генах. Значительное число мутаций в различных генах было описано как патогенные для Ph-негативных МПН, оказывающие влияние на фенотип, течение и прогноз заболевания. Быстрое и достоверное выявление таких мутаций в широком спектре генов невозможно рутинными лабораторными методами. В этой связи все большее применение находит метод NGS, который позволяет с высокой точностью и чувствительностью одновременно определить мутации в большом количестве генов.
В настоящий момент как в научной, так и клинической практике диагностику пациентов с Ph-негативными МПН методом NGS проводят с использованием различных индивидуальных и коммерческих панелей генов [9–12]. В нашем исследовании впервые мутационный статус пациентов с Ph-негативными МПН изучен с помощью персонализированной панели из 118 генов. Данная панель включает в себя не только гены, ассоциированные с Ph-негативными МПН, но и гены, мутации в которых выявляются при других типах миелоидных новообразований. Секвенирование было проведено у 83 Ph-негативных пациентов с МПН. Анализ данных показал, что мутации в генах JAK2, MPL и CALR, выявленные у пациентов стандартными лабораторными методами, успешно обнаруживаются в наших условиях методом NGS. Этот факт свидетельствует о надежности полученных данных секвенирования.
В нашем исследовании в дополнение к драйверным генам мутации, определяемые как патогенные, были выявлены у 39 пациентов в 23 (20%) генах из 118 исследуемых. По сравнению с результатами, полученными в исследовании авторов [13], при анализе группы из 197 пациентов методом NGS с использованием таргетной панели патогенные мутации были найдены у 35% больных. При этом мутации были выявлены в 27% генов из 104 анализируемых. Различия в количестве мутировавших генов (20% и 27%) от общего их числа можно объяснить разным набором генов, используемых в таргетных панелях в нашей работе и исследованиях Lundberg et al. [13].
Наиболее часто дополнительные патогенные мутации у пациентов с различными МПН встречаются в гене ASXL1 [8]. Мы обнаружили, что 25% пациентов имели мутацию именно в данном гене. Частота выявления мутаций в гене ASXL1 варьирует при различных Ph-негативных МПН. Так, по данным исследователей [14–19], при первичном миелофиброзе мутации были обнаружены в 23–25% случаев, при эссенциальной тромбоцитемии — в 5–20%, при истинной полицитемии частота мутаций в гене ASXL1 составила 3,5–11,8%. В нашем исследовании данные мутации были обнаружены у 26,6% пациентов с ПМФ, у 14,2% больных с ЭТ и 16% пациентов с ИП.
При клональной эволюции МПН предполагаются два пути мутагенеза с участием гена ASXL1:
Многие исследования показывают влияние мутаций в гене ASXL1 на общую и бессобытийную выживаемости и тромботические события у пациентов с Ph-негативными МПН [23][24]. Однако Guglielmelli et al. установили, что мутации в гене ASXL1 снижают общую выживаемость у пациентов с ПМФ, но не с вторичным МФ [25]. Кроме того, мутации в ASXL1 оказывают влияние на результаты терапии таргетными препаратами и алло-ТГСК [26]. В нашем исследовании мы показали ассоциацию драматического снижения бессобытийной выживаемости (медиана выживаемости 7,83 против 15,75 года, р = 0,0011) для всех пациентов с мутациями и без в данном гене.
Достижения генетики и молекулярной биологии последних лет позволили более полно описать генетический ландшафт Ph-негативных МПН и выявить гены, мутации в которых оказывают негативное влияние на прогноз: ASXL1, EZH2, IDH1, IDH2, SRSF2 и U2AF1Q157. Такие мутации получили название «мутации высокого риска» и были включены в различные прогностические шкалы. Количество мутаций в данных генах также влияет на прогноз, а именно: 1 патогенная мутация сокращала медиану общей выживаемости больных в 1,7 раза, 2 мутации — в 4,7 раза по сравнению с пациентами без мутаций в этих генах (Me = 12,2, Me = 7,0 , Me = 2,6 года соответственно, p < 0,0001) для пациентов с миелофиброзом [27].
В нашем исследовании с использованием панели из 118 генов мы показали, что не только мутации высокого риска оказывали влияние на выживаемость пациентов с МПН, но и любые патогенные варианты мутаций в сочетании с драйверными генами (p = 0,0004) существенно снижали ее; при этом важным является не только сам факт наличия данного варианта сочетания, но и количество таких вариантов. У пациентов с двумя и более мутациями значительно снижалась бессобытийная выживаемость по сравнению с пациентами с одной мутацией (p < 0,0001). Таким образом, при диагностике Ph-негативного МПН важно не ограничиваться определением мутационного статуса только 6 генов высокого молекулярного риска, но анализировать как можно более широкие панели.
Недавние исследования продемонстрировали важность NGS метода для описания мутационного профиля пациентов с тройным негативным статусом [28][29]. Действительно, у 8 из 10 таких пациентов из изучаемой нами когорты были выявлены патогенные мутации в различных генах. Обнаружение мутаций у данной группы пациентов позволило подтвердить клональность и оценить риски течения заболевания. Важно отметить, что отсутствие драйверных и патогенных мутаций у пациентов с подтвержденным диагнозом Ph-негативного МПН позволяет выделить их в отдельную когорту с наиболее благоприятным прогнозом течения заболевания без лейкемической трансформации [12]. Не во всех исследованиях, однако, удается выделить такую группу. Так, Huang et al. выявили патогенные мутации у всех 12 пациентов с тринегативным статусом [12].
Метод NGS является удобным инструментом как для оценки прогноза заболевания, так и для выбора генов-мишеней таргетной терапии, направленной не только на драйверные, но и на гены с различным функционалом (например, IDH1,2 и EZH2). Мутации в данных генах были выявлены и у пациентов из нашей когорты. При этом гены IDH1,2 были мутированы у 8% пациентов, а EZH2 — у 3% пациентов, что соответствует данным, полученным другими исследователями с помощью стандартных молекулярных методов [8][30].
Соматические мутации в широком спектре генов выявляются у 80% пациентов с ПМФ и у 50% пациентов с ЭТ/ИП и оказывают влияние на течение и прогноз заболевания [29]. В нашей группе пациентов мы обнаружили патогенные мутации, влияющие на прогноз течения заболеваний, у 46% больных с ПМФ, в том числе и у пациентов с лейкемической трансформацией. К факторам риска, повышающим вероятность лейкемической трансформации, среди прочего, относят мутации в различных генах: IDH1, IDH2, SRSF2, ASXL1 при первичном миелофиброзе, в генах SRSF2, IDH2 или RUNX1 при истинной полицитемии, в генах TP53, SRSF2, EZH2, U2AF1 или RUNX1 при эссенциальной тромбоцитемии. При этом было показано, что время до лейкемической трансформации сокращается с увеличением количества патогенных мутаций у пациентов с Ph-негативными МПН (p < 0,0001), что коррелирует с полученными нами данными о большем количестве мутаций у пациентов с трансформацией заболеваний [12].
Обнаружение драйверных мутаций стало прорывным открытием в диагностике миелопролиферативных новообразований, что помогло в определении патогенеза этих заболеваний. Сейчас внедрение метода NGS значительно изменяет восприятие и подход к диагностике, оценке риска и лечению пациентов с Ph-негативными МПН. Благодаря возможности одновременного поиска мутаций во многих генах NGS позволяет не только устанавливать диагноз и подтверждать клональность заболевания, но и выявлять группы пациентов с неблагоприятным прогнозом и повышенным риском прогрессирования и трансформации заболевания.
Таким образом, применение метода NGS с использованием панели из 118 генов при диагностике больных Ph-негативными миелопролиферативными новообразованиями позволило изучить мутационный профиль заболевания, подтвердить клональность заболевания у пациентов с тринегативным статусом, выявить патогенные мутации, значимо влияющие на результаты терапии пациентов.
В нашем исследовании патогенные мутации выявлялись практически у половины пациентов с Ph-негативными МПН в 23 генах. Снижение бессобытийной выживаемости было показано для больных с сочетанием драйверной и патогенной мутаций. Две и более патогенные мутации у одного пациента сокращали бессобытийную выживаемость по сравнению с пациентами с одной мутацией. Наиболее частым молекулярным событием при Ph-негативных МПН являлись мутации в гене ASXL1, ассоциирующиеся со снижением бессобытийной выживаемости больных. Комплексный подход к диагностике Ph-МПН с применением современных молекулярно-генетических технологий позволит устанавливать диагноз, оценивать прогностические особенности течения заболевания и подбирать наиболее эффективную персонализированную терапию.
1. Шуваев ВА, Мартынкевич ИС, Сидоркевич СВ. Миелопролиферативные новообразования. М.: Буки Веди; 2023.
2. Меликян АЛ, Ковригина АМ, Суборцева ИН. Шуваев ВА, Морозова ЕВ, Ломаиа ЕГ и др. Национальные клинические рекомендации по диагностике и лечению Ph-негативных миелопролиферативных заболеваний (истинной полицитемии, эссенциальной тромбоцитемии, первичного миелофиброза) (редакция 2020 г.). Клиническая онкогематология. 2021;14(2):262–98. https://doi.org/10.21320/2500-2139-2021-14-2-262-298
3. Spivak JL. Myeloproliferative Neoplasms. N. Engl. J. Med. 2017;376:2168–81. https://doi.org/10.1056/NEJMra1406186
4. Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF. et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022;36:1703–19. https://doi.org/10.1038/s41375-022-01613-1
5. Hu X, Li J, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther. 2021;6(1):402. https://doi.org/10.1038/s41392-021-00791-1
6. Tefferi A, Lasho TL, Finke CM, Elala Y, Hanson CA, Ketterling RP et al. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016;30:105–11. https://doi.org/10.1182/bloodadvances.2016000208
7. Tefferi A, Lasho TL, Guglielmelli P, Finke CM, Rotunno G, Elala Y. et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016;22:21–30. https://doi.org/10.1182/bloodadvances.2016000216
8. Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood. 2017;129(6):667–79. https://doi.org/10.1182/blood-2016-10-695940
9. Zuo Z, Li S, Xu J, You MJ, Khoury JD, Yin CC. Philadelphia-Negative Myeloproliferative Neoplasms: Laboratory Workup in the Era of Next-Generation Sequencing. Curr Hematol Malig Rep. 2019;14(5):376–85. https://doi.org/10.1007/s11899-019-00534-8
10. Alduaij W, McNamara CJ, Schuh A, Arruda A, Sukhai M, Kanwar N. et al. Clinical Utility of Next-generation Sequencing in the Management of Myeloproliferative Neoplasms: A Single-Center Experience. Hemasphere. 2018;2(3):e44. https://doi.org/10.1097/HS9.0000000000000044
11. Visani G, Etebari M, Fuligni F, Di Guardo A, Isidori A, Loscocco F. et al. Use of Next Generation Sequencing to Define the Origin of Primary Myelofibrosis. Cancers (Basel). 2023;15(6):1785. https://doi.org/10.3390/cancers15061785
12. Huang X, Wu J, Deng X, Xu X, Zhang X, Ma W. et al. Mutation profiles of classic myeloproliferative neoplasms detected by a customized next-generation sequencing-based 50-gene panel. Journal of Bio-X Research .2020;3(1):13–20. https://doi.org/10.1097/JBR.0000000000000061
13. Lundberg P, Karow A, Nienhold R, Looser R, Hao-Shen H, Nissen I. et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood. 2014;123(14):2220–8. https://doi.org/10.1182/blood-2013-11-537167
14. Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12:599–612. https://doi.org/10.1038/nrc3343
15. Viny AD, Levine RL. Genetics of myeloproliferative neoplasms. Cancer J. 2014;20:61–5. https://doi.org/10.1016/j.hoc.2020.12.002
16. Milosevic JD, Kralovics R. Genetic and epigenetic alterations of myeloproliferative disorders. Int J Hematol. 2013;97:183–97. https://doi.org/10.1007/s12185-012-1235-2
17. Guglielmelli P, Gangat N, Coltro G, Lasho TL, Loscocco GG, Finke CM. et al. Mutations and thrombosis in essential thrombocythemia. Blood Cancer J. 2021;11(4):77. https://doi.org/10.1038/s41408-021-00470-y
18. Segura-Díaz A, Stuckey R, Florido Y, Sobas M, Álvarez-Larrán A, Ferrer-Marín F. et al. DNMT3A/TET2/ASXL1 mutations are an age-independent thrombotic risk factor in polycythemia vera patients: an observational study. Thromb Haemost. 2024;124(7):669–75. https://doi.org/10.1055/a-2239-9265
19. Fujino T, Kitamura T. ASXL1 mutation in clonal hematopoiesis. Exp Hematol. 2020;83:74–84. https://doi.org/10.1016/j.exphem.2020.01.002
20. Gelsi-Boyer V, Brecqueville M, Devillier R, Murati A, Mozziconacci MJ, Birnbaum D. Mutations in ASXL1 are associated with poor prognosis across the spectrum of malignant myeloid diseases. J Hematol Oncol. 2012;5:12. https://doi.org/10.1186/1756-8722-5-12
21. Triviai I, Zeschke S, Rentel J, Spanakis M, Scherer T, Gabdoulline R. et al. ASXL1/EZH2 mutations promote clonal expansion of neoplastic HSC and impair erythropoiesis in PMF. Leukemia. 2019;33(1):99–109. https://doi.org/10.1038/s41375-018-0159-0
22. Svidnicki C, M., De Melo Campos P., Filho A.F., Fujiura L.C., Yoshizato T., Makishima H., et al. Mutations in triple-negative patients with myeloproliferative neoplasms. Blood. 2019;134(1):5395. https://doi.org/10.1182/blood-2019-128764
23. Regimbeau M, Mary R, Hermetet F, Girodon F. Genetic Background of Polycythemia Vera. Genes. 2022;13(4):637. https://doi.org/10.3390/genes13040637
24. Nie YB, Sun M, He CK, Ju MK, Zhou FL, Wu SY. et al. ASXL1 mutations in Chinese patients with essential thrombocythemia. Exp Ther Med. 2015;15(5):4149–56. https://doi.org/10.3892/etm.2018.5939
25. Guglielmelli P, Coltro G, Mannelli F, Rotunno G, Loscocco GG, Mannarelli C. et al. ASXL1 mutations are prognostically significant in PMF, but not MF following essential thrombocythemia or polycythemia vera. Blood Adv. 2022;6(9):2927–31. https://doi.org/10.1182/bloodadvances.2021006350
26. Spiegel J, McNamara C, Kennedy J, et al. lmpact of genomic alterations on outcomes in myelofibrosis patients undergoing JAKl/2 inhibitor therapy. Blood Adv.2017;1(20):1729–38. https://doi.org/10.1182/bloodadvances.2017009530
27. Guglielmelli P, Lasho TL, Rotunno G, Score J, Mannarelli C, Pancrazzi A. et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: an international study of 797 patients. Leukemia. 2014;28(9):1804–10. https://doi.org/10.1038/leu.2014.76
28. Wu S, Luo P, Yu Y, Xiong B, Wang Y, Zuo X. Next-generation sequencing redefines the diagnosis of triple-negative myeloproliferative neoplasms. Ann Hematol. 2022;101(3):705–8. https://doi.org/10.1007/s00277-021-04561-5
29. Mroczkowska-Bękarciak A, Wróbel T. BCR::ABL1-negative myeloproliferative neoplasms in the era of next-generation sequencing. Front Genet. 2023;14:1241912. https://doi.org/10.3389/fgene.2023.1241912
30. Shih A, Abdel-Wahab O, Patel J, Levine R. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12(9):599–612. https://doi.org/10.1038/nrc3343
Кириенко Анна Николаевна - канд. биол. наук.
Санкт-Петербург
Мотыко Екатерина Вадимовна - канд. биол. наук
Санкт-Петербург
Ефремова Елизавета Викторовна
Санкт-Петербург
Кустова Дарья Владимировна
Санкт-Петербург
Герт Татьяна Николаевна
Санкт-Петербург
Леппянен Ирина Викторовна - канд. биол. наук
Санкт-Петербург
Шуваев Василий Анатольевич - д-р мед. наук
Москва; Обнинск
Мартынкевич Ирина Степановна - д-р мед. наук
Санкт-Петербург
Кириенко А.Н., Мотыко Е.В., Ефремова Е.В., Кустова Д.В., Герт Т.Н., Леппянен И.В., Шуваев В.А., Мартынкевич И.С. Изучение мутационного профиля больных Ph-негативными миелопролиферативными новообразованиями методом NGS. Экстремальная биомедицина. 2025;27(1):80-87. https://doi.org/10.47183/mes.2024-241
Kirienko A.N., Motyko E.V., Efremova E.V., Kustova D.V., Gert T.N., Leppyanen I.V., Shuvaev V.A., Martynkevich I.S. NGS analysis of the mutational profile of patients with Ph-negative myeloproliferative neoplasms. Extreme Medicine. 2025;27(1):80-87. https://doi.org/10.47183/mes.2024-241
119121, Москва, Погодинская ул., д. 10, стр. 1
Тел.: +7 (495) 540-61-71, доб.: 4190, 4191, 4192
E-mail: extrememedicine@cspfmba.ru












