



Contents
Scroll to:
https://doi.org/10.47183/mes.2025-284
Scroll to:
Introduction. Primary immunodeficiency disorders (PID) are a group of congenital diseases caused by genetic defects that lead to diverse phenotypic manifestations. Classic inflammatory bowel diseases (IBD) are typically multifactorial pathologies, combining genetic predisposition, gut microbiota alterations, and adverse environmental influences. Very early-onset IBD (VEO-IBD), defined as a disease presenting before six years of age, accounts for 3–15% of all pediatric IBD. This subgroup is particularly characterized by monogenic etiology, associated with gastrointestinal phenotype PID and with causative mutations in specific genes.
Case report. We present a clinical case of monogenic Crohn’s disease in a child with X-linked lymphoproliferative syndrome type 2 (XLP-2), treated using a multi-stage team approach. To achieve sustained remission, in addition to selecting conservative therapy, repeated surgical interventions and repeated hematopoietic stem cell transplantations were required. An individualized approach and treatment strategy planning enabled a positive treatment outcome.
Conclusions. In young children with the presence of atypical IBD and refractoriness to standard therapy, it is crucial to differentiate monogenic forms of IBD. Such patients require close monitoring, dynamic follow-up, and a multidisciplinary approach involving collaboration between gastroenterologists, immunologists, and surgeons.
Alieva E.I., Shcherbakova O.V., Zyabkin I.V. Crohn’s disease complicating a rare primary immunodeficiency in a pediatric patient: challenges in medical and surgical decision-making. Extreme Medicine. 2025;27(3):410-416. https://doi.org/10.47183/mes.2025-284
Primary immunodeficiency disorders (PID) are genetically determined diseases characterized by congenital defects in the body’s defense mechanisms, leading to impaired immune responses with the development of recurrent infections, increased risk of malignancies, and autoimmune diseases [1]. The current classification of PID adopted by the International Union of Immunological Societies (IUIS) takes into account molecular genetic defects in various immunodeficiencies, identifying 10 main groups of PID [2].
X-linked lymphoproliferative syndrome (XLP) belongs to the group of PID with immune dysregulation; XLP is caused by mutations in the SH2D1A, XIAP, and MAGT1 genes, characterized by an atypical response to Epstein–Barr virus infection and the development of hemophagocytosis, dysgammaglobulinemia, and malignant lymphoproliferation. The former two types (XLP1 and XLP2) are among the most studied types, which occur with a frequency of 1–3 per 1 million born boys, generating interest given the rarity of this disease. A significant difference between XLP2 and XLP1 is the development of hemorrhagic colitis, which clinically and morphologically resembles inflammatory bowel disease (IBD).
IBDs are classified as polygenic inherited disorders, with more than a hundred susceptibility loci that increase the risk of the disease having been identified. To date, over 300 , mutations in which lead to the development of PID genes, have been studied [3]. Among these, more than 75 genes whose mutations are associated with an IBD-like phenotype have been found [4]. In children with very early-onset IBD (VEO IBD) (before 6 years of age) and especially infantile IBD (with the disease onset before 2 years of age), differential diagnosis with PID is necessary [5]. IBD in the setting of a monogenic disease almost always follows a course similar to classical multifactorial IBD, differing in refractoriness to therapy. This may be the sole manifestation of PID at the time of presentation, delaying the verification of a final diagnosis [6]. The main treatment methods for PID include conservative therapy (symptomatic, antibacterial, and antifungal), immunoglobulin replacement therapy, treatment with genetically engineered biological drugs (GEBD) and targeted immunosuppressive agents, and in certain cases, hematopoietic stem cell transplantation (HSCT) [7].
However, to date, versatile approaches to treating such patients have been lacking. An important separate treatment option for patients with PID involving the colon is minimally invasive surgical interventions, such as preventive ileostomy before HSCT to reduce the risk of transplantation complications [8].
We present a clinical case of a pediatric patient with PID and Crohn’s-like intestinal involvement, discussing the challenges of therapeutic and surgical treatment stages.
Patient B., male, 8 years old (born in 2016), was under observation at the gastroenterology department of the Federal Research and Clinical Center for Children and Adolescents of FMBA with the following diagnoses:
The child was born from the 2nd physiological pregnancy, 2nd term spontaneous delivery (1st child — girl, 10 years old — healthy). Birth length was 49 cm, and birth weight was 2860 g. The Apgar score was 8/8. The child was breastfed from the first day, continued breastfeeding until 1.5 years old. BCG (Calmette-Guerin bacillus) vaccinated at birth. The history of infections: frequent ARVI (at 1 month — ARVI with respiratory failure), pneumonia at 1 year. Routine vaccinations according to national schedule (medical exemption since 07.2017). Diaskintest (29.08.2017) was negative. No significant family history.
From 3 weeks of age, the child had frequent loose stools with mucus, and from 9 months — blood streaks in stool. At 1 year old, the child developed pneumonia followed by several months of febrile episodes. In May 2017 (1 year 4 months), surgical treatment for acute paraproctitis (abscess incision and drainage) was performed.
Clinical deterioration in July 2017 led to hospitalization at the gastroenterology department of Morozov Children’s City Clinical Hospital. On examination: fistulous opening with purulent discharge in the anal area, perianal dermatitis with maceration, anal fissure (Fig. 1).
Laboratory findings revealed: leukocytosis 22.3 × 103 cells/μL, decreased hemoglobin to 57 g/L, elevated C-reactive protein (CRP) to 64.9 mg/L, and immunogram abnormalities (leukocyte differentiation cluster — CD3+ 2000/μL, CD4+ 670/μL, CD8+ 1335/μL, CD19+ 339/μL, NK cells 55/μL). During hospitalization at the gastroenterology department in August 2017, an axillary lymphadenitis episode was noted, requiring incision and drainage of the abscess.
Colonoscopy identified ulcerative lesions of the colon, while histological examination revealed features of chronic left-sided ulcerative colitis with high activity. In 2017, gastroenterologists determined the diagnosis: Crohn’s disease affecting the colon, with suspected primary immunodeficiency. Treatment was initiated with prednisone and 5-ASA (5-amino salicylic acid) drugs, supplemented by antimicrobial and antifungal therapy (amoxicillin + clavulanic acid, ciprofloxacin, cefoperazone + sulbactam, fluconazole). The child was referred for genetic testing (whole-exome genome sequencing).
In February 2018, molecular genetic testing confirmed a pathogenic variant in exon 2 of the XIAP gene: c.599G > A (p.Cys200Tyr) in hemizygous state. In March 2018, due to progressive deterioration marked by severe colitis syndrome, the child was admitted to the immunology department of Dmitry Rogachev National Research Center.
The patient was subscribed:
A medical consensus recommended diverting ileostomy prior to hematopoietic stem cell transplantation (HSCT) to reduce complications. Preoperative stoma site marking (Fig. 2) and laparoscopic-assisted double-barrel ileostomy (01.06.2018) was performed.
Laparoscopic examination of the abdominal cavity revealed the following findings: the right colon had normal wall thickness but was dilated to 4 cm in diameter; the sigmoid colon showed marked inflammatory changes over an 8–10 cm segment with thickened walls and creeping mesentery. The postoperative course was uneventful.
Subsequent treatment was conducted at the Dmitry Rogachev National Research Center. An allogeneic hematopoietic stem cell transplantation from a related haploidentical donor (mother) with TCRαβ/CD19 depletion was performed on 25.07.2018. On 15.08.2018, a complication occurred, involving ultra-early graft rejection.
Given the graft rejection, severe enteritis, and high ileostomy output (laboratory tests showed no infectious process with restored native leukopoiesis), therapy with recombinant monoclonal antibodies adalimumab 40 mg every 2 weeks was reinitiated.
A second allogeneic hematopoietic stem cell transplantation from a haploidentical donor (father) with TCRαβ/CD19-depleted graft processing was performed on 29.11.2018. Autoimmune hemolysis developed in early May 2019 (anemia with hemoglobin dropping to 73 g/L, reticulocytes 10%, direct Coombs test ++++). The child showed a progressive shift toward host chimerism and developed autoimmune hemolysis refractory to IV immunoglobulin, prompting initiation of rituximab therapy at an initial dose of 375 mg/m2 with positive effect.
In August 2019, the child was admitted to the Dmitry Rogachev National Research Center with severe pain and bowel evagination through the stoma. Conservative reduction attempts under tramadol analgesia and mask anesthesia were unsuccessful. Necrosis of the evaginated bowel was diagnosed, and emergency surgery was performed: bowel resection with creation of a double-barreled divided ileostomy. Postoperative wound dehiscence required secondary surgical wound debridement and stoma revision.
In September 2019, the child was admitted to the surgical department of the Russian Children’s Clinical Hospital with peristomal complications (Fig. 3). Stoma therapy was performed with positive effect. Endoscopic examination of the excluded colon (Fig. 4) revealed erosive proctosigmoiditis and sigmoid colon stricture with lumen obliteration.
On 01.11.2019, at the Dmitry Rogachev National Research Center, graft rejection was confirmed based on bone marrow chimerism analysis (93.7% host cells). A discussion was held with the patient’s parents regarding the need for repeat hematopoietic stem cell transplantation and associated risks. Given persistent erosive changes and stricture development in the excluded colon segments, the patient continued local therapy with Salofalk, adalimumab therapy at a reduced frequency of 40 mg weekly, initiated immunosuppressive therapy with azathioprine, and maintained complex antimicrobial therapy with intravenous immunoglobulin replacement. Repeat HSCT was declined due to autoimmune hemolytic anemia, granulocytic graft hypofunction, and severe colitis syndrome.
Considering the extremely high risk of worsening gastrointestinal damage from diversion colitis in the excluded intestinal segments, the decision was made to perform reconstructive surgery with resection of the strictured bowel segment, simultaneous ileostomy closure, and intestinal continuity restoration.
On 27.11.2019, at the Russian Children’s Clinical Hospital (RCCH), the following elective procedures were performed: resection of the sigmoid colon stricture with colocolic anastomosis; ileostomy closure with ileocolonic anastomosis. Intraoperative examination revealed the entire colon to appear thickened, reduced in diameter (typical “excluded bowel” appearance), with markedly thickened mesentery and serosal surface nearly obscured by “creeping fat” (Fig. 5). A 4 cm segment of significant luminal narrowing was identified in the sigmoid colon. The efferent ileostomy limb was located 2 cm from the ileocecal valve, with the appendix embedded in adhesions between the afferent and efferent limbs. Resection of the ileocecal region was performed with end-to-end ileoascending anastomosis (Fig. 6).
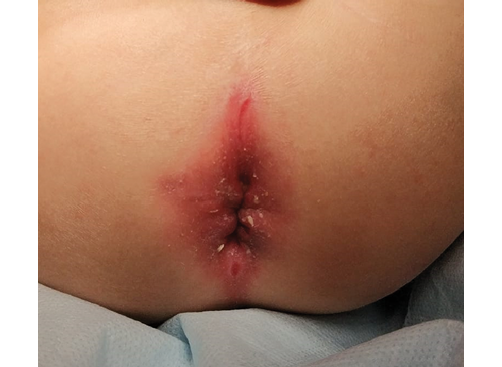
Clinical photo taken by the authors
Fig. 1. Perianal region appearance. Multiple anal fissures and perianal dermatitis were observed

Clinical photo taken by the authors
Fig. 2. Preoperative stoma site marking
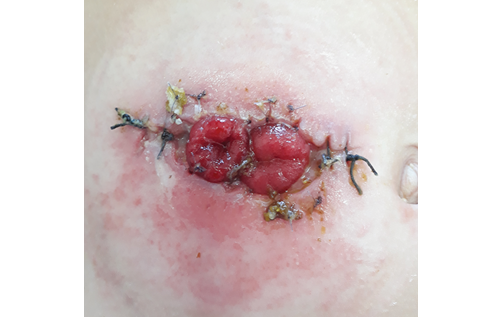
Clinical photo taken by the authors
Fig. 3. Peristomal complications: infected wound edges and dermatitis around the stoma
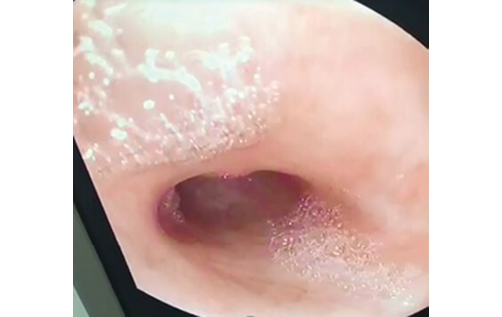
Clinical photo taken by the authors
Fig. 4. Endoscopic image (sigmoidoscopy data). Obliteration of the sigmoid colon lumen visualized endoscopically
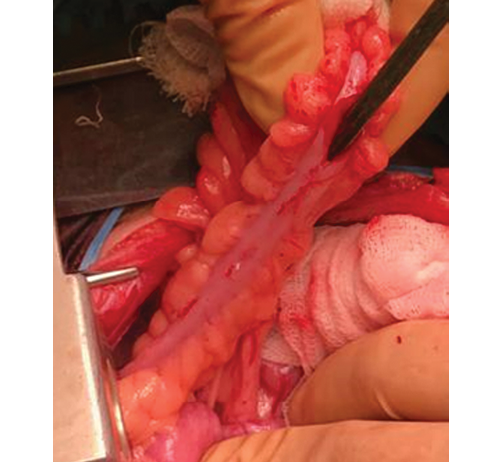
Clinical photo taken by the authors
Fig. 5. Intraoperative photograph. View of the sigmoid colon almost obscured by “creeping fat”: grooved probe inserted into the lumen up to the stricture
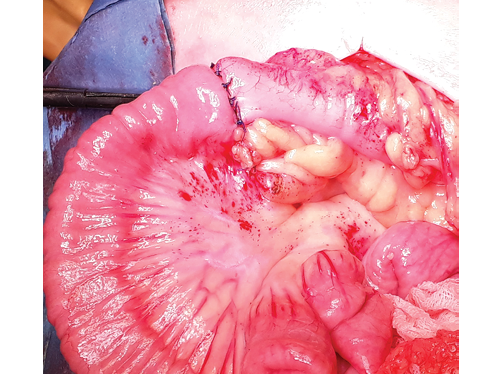
Clinical photo taken by the authors
Fig. 6. Intraoperative photograph. View of the direct ileo-ascendoanastomosis
Histopathological examination revealed:
The child’s condition remained stable, with persistent host-dominant chimerism (93.7% host cells). During interhospital periods, biologic therapy with adalimumab 40 mg every 2 weeks was maintained. Immunosuppressive therapy (azathioprine) was discontinued in August 2020.
Since August 2021, the patient has been followed at the gastroenterology department of the Federal Research and Clinical Center for Children and Adolescents, undergoing regular medical examinations. Sustained clinical-endoscopic remission has been maintained. Control endoscopic examinations show:
In January 2022, the examination at Zurich Children’s Hospital (Switzerland) recommended:
The current management includes:
Very early-onset IBD (VEO-IBD) is a rare condition characterized by the disease onset before 6 years of age, occurring in 3–15% of pediatric IBD cases. It presents with severe clinical activity, refractoriness to therapy, and higher mortality rates [5][9]. Conventional treatments for early-onset IBD, such as anti-TNF therapy, demonstrate limited efficacy [10]. In cases of VEO-IBD (particularly before 2 years of age) or when primary immunodeficiency (PID) warning signs are present [4][7], diagnostic evaluation for monogenic mutations with PID verification should be performed [11].
We present a clinical case of monogenic Crohn’s disease manifesting with intestinal symptoms since the neonatal period, perianal lesions, recurrent infections of various localizations in early childhood, atypical disease course, and refractoriness to standard medical therapy. The gastroenterologist awareness and vigilance enabled timely suspicion of PID and subsequent whole-exome sequencing, which revealed X-linked lymphoproliferative syndrome type 2 (XLP).
Among all PIDs, XLP2 most frequently exhibits IBD-like clinical presentation, with colonic involvement mimicking Crohn’s disease occurring in 19% of children with XLP2 [12]. XLP results from mutations in the XIAP gene (X-linked inhibitor of apoptosis protein), also known as BIRC4 (baculoviral IAP repeat-containing protein 4), leading to enhanced apoptosis of regulatory T-lymphocytes (NKT cells and CD4+ Tregs) and consequent marked pathogen-induced immune responses [13].
Beyond timely PID diagnosis, a key factor in successful management of monogenic Crohn’s disease is surgical intervention with diverting stoma formation, aimed at controlling colonic inflammation and improving nutritional status. Currently, there are no international or national guidelines on indications for surgical intervention in refractory colitis, with only isolated publications supporting ileostomy creation to alleviate severe clinical symptoms in children with VEO-IBD and to enable stem cell transplantation [8][14]. We concur with international researchers regarding the importance of preventive ileostomy prior to HSCT to reduce potential complications and advocate for fecal diversion as part of staged management for severe refractory colitis with perianal involvement in Crohn’s disease and Crohn’s-like PID.
In our case, we demonstrated that elective preventive ileostomy ensured control of septic (infectious) complications and reduced intestinal risks during HSCT, despite episodes of graft rejection.
While stoma may temporarily alleviate symptoms, it cannot ensure long-term clinical remission in children with IBD. Moreover, the incidence of various peristomal complications increases over time [15]. In our case, we observed such complications one year after stoma formation (stoma evagination, bowel necrosis, and wound dehiscence following emergency surgery). Additionally, prolonged ileostomy with excluded colon may lead to “diversion colitis,” requiring differentiation from immune-mediated colitis flare when treatment refractoriness develops. The etiology of the sigmoid colon stricture remains unclear — it could have resulted from chronic graft-versus-host reaction or Crohn’s-like transmural colonic involvement.
Despite initial inefficacy of adalimumab and unsuccessful repeat HSCT, an individualized approach combining optimized medical therapy with symptomatic treatment and well-timed staged surgical interventions allowed us to achieve favorable outcomes in this child with severe PID manifesting Crohn’s-like intestinal and perianal involvement.
Timely diagnosis of primary immunodeficiency (PID) enables prompt initiation of targeted therapy or hematopoietic stem cell transplantation (HSCT), which in most cases leads to disease remission and improves patients’ quality and duration of life.
Optimal patient-centered management strategies for children with severe conditions like monogenic forms of inflammatory bowel disease (IBD) require:
1. Yu JE, Orange JS, Demirdag YY. New primary immunodeficiency diseases: context and future. Current Opinion in Pediatrics. 2018;30(6):806–20. https://doi.org/10.1097/MOP.0000000000000699
2. Bousfiha A, Moundir A, Tangye SG, Picard C, Jeddane L, Al-Herz W, et al. The 2022 Update of IUIS Phenotypical Classification for Human Inborn Errors of Immunity. Journal of Clinical Immunology. 2022;42(7):1508–20. https://doi.org/10.1007/s10875-022-01352-z
3. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the International Union of Immunological Societies Expert Committee. Journal of Clinical Immunology. 2022;42(7):1473–507. https://doi.org/10.1007/s10875-022-01289-3
4. Uhlig HH, Charbit-Henrion F, Kotlarz D, Shouval DS, Schwerd T, Strisciuglio C, et al. Clinical genomics for the diagnosis of monogenic forms of inflammatory bowel Disease: A position paper from the Paediatric IBD Porto Group of European Society of Paediatric Gastroenterology, Hepatology and Nutrition. Journal of Pediatric Gastroenterology and Nutrition. 2021;72(3):456–73. https://doi.org/10.1097/MPG.0000000000003017
5. Shchigoleva АЕ, Shumilov PV, Suspitsin ЕN. The role of targeted sequencing in diagnosis of very-early-onset inflammatory bowel disease. Pediatric Nutrition. 2020;18(1):28–34 (In Russ.). https://doi.org/10.20953/1727-5784-2020-1-28-34
6. Tegtmeyer D, Seidl M, Gerner P, Baumann U, Klemann C. Inflammatory bowel disease caused by primary immunodeficiencies — Clinical presentations, review of literature, and proposal of a rational diagnostic algorithm. Pediatric Allergy and Immunology. 2017;28(5):412–29. https://doi.org/10.1111/pai.12734
7. Shcherbina AYu. Masks of primary immunodeficiency disorders: diagnostic and therapeutic problems. Russian Journal of Pediatric Hematology and Oncology. 2016;3(1):52–8 (In Russ.). https://doi.org/10.17650/2311-1267-2016-3-1-52-58
8. Sun S, Ye Z, Zheng S, Chen G, Qian X, Dong K, et al. Surgical treatment of monogenic infammatory bowel disease: A single clinical center experience. Journal of Pediatric Surgery. 2019;54(10):2155–61. https://doi.org/10.1016/j.jpedsurg.2019.02.013
9. Kelsen JR, Sullivan KE, Rabizadeh S, Singh N, Snapper S, Elkadri A, et al. North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition position paper on the evaluation and management for patients with very early-onset inflammatory bowel disease. Journal of Pediatric Gastroenterology and Nutrition. 2020;70(3):389–403. https://doi.org/10.1097/MPG.0000000000002567
10. Bramuzzo M, Arrigo S, Romano C, Filardi MC, Lionetti P, Agrusti A, et al. Efficacy and safety of infliximab in very early onset inflammatory bowel disease: a national comparative retrospective study. United European Gastroenterology Journal. 2019;7(6):759–66. https://doi.org/10.1177/2050640619847592
11. Ouahed J, Spencer E, Kotlarz D, Shouval DS, Kowalik M, Peng K, et al. Very early onset inflammatory bowel disease: A clinical approach with a focus on the role of genetics and underlying immune deficiencies. Inflammatory Bowel Diseases. 2020;26(6):820–42. https://doi.org/10.1093/ibd/izz259
12. Balashov DN, Roppelt AA, Rumjantsev AG, Shcherbina AU. Х-linked lymphoproliferative syndrome. Federal clinical recommendations, 2020. Russian Journal of Allergology. 2019;16(4):66–77 (In Russ.). https://doi.org/10.36691/RAJ.2020.16.4.008
13. Aguilar C, Latour S. X-linked inhibitor of apoptosis protein deficiency: more than an X-linked lymphoproliferative syndrome. Journal of Clinical Immunology. 2015;35(4):331–8. https://doi.org/10.1007/s10875-015-0141-9
14. Maxwell EC, Dawany N, Baldassano RN, Mamula P, Mattei P, Albenberg L, et al. Diverting ileostomy for the treatment of severe, refractory, pediatric inflammatory bowel disease. Journal of Pediatric Gastroenterology and Nutrition. 2017;65(3):299–305. https://doi.org/10.1097/MPG.0000000000001498
15. Xu X, Zhou Y, Tan Z, Huang Y, Dong K, Gu Y, et al. Risk factors for stoma and incision complications of enterostomy in children with very early-onset inflammatory bowel disease: a prospective cohort study. European Journal of Pediatrics. 2025;184(2):146–70. https://doi.org/10.1007/s00431-024-05952-2
16.
Elmira I. Alieva, Dr. Sci. (Med.)
Moscow
Olga V. Shcherbakova, Dr. Sci. (Med.)
Moscow
Ilya V. Zyabkin, Dr. Sci. (Med.)
Moscow
Alieva E.I., Shcherbakova O.V., Zyabkin I.V. Crohn’s disease complicating a rare primary immunodeficiency in a pediatric patient: challenges in medical and surgical decision-making. Extreme Medicine. 2025;27(3):410-416. https://doi.org/10.47183/mes.2025-284
10 bld. 1 Pogodinskaya Str., Moscow, Russia 119121
tel.: +7 (495) 540-61-71, ext.: 4190, 4191, 4192
E-mail: Extrememedicine@cspfmba.ru
Processing of personal data












