


Содержание
Перейти к:
 Е. Н. Черняева,
K. В. Морозов,
A. Д. Мацвай,
M. С. Гуськова,
А. Ю. Некрасов,
И. Ф. Стеценко,
А. О. Носова,
О. Г. Курская,
А. М. Шестопалов,
Г. A. Шипулин
Е. Н. Черняева,
K. В. Морозов,
A. Д. Мацвай,
M. С. Гуськова,
А. Ю. Некрасов,
И. Ф. Стеценко,
А. О. Носова,
О. Г. Курская,
А. М. Шестопалов,
Г. A. Шипулин https://doi.org/10.47183/mes.2024-26-3-40-50
Перейти к:
Введение. Молекулярно-генетическое исследование штаммов Measles morbillivirus, позволяющее оценивать гетерогенность клинически значимых штаммов, определять источники заражения и пути передачи инфекции, в настоящее время является актуальной задачей. В последние два года наблюдается рост заболеваемости корью в России и других странах. В данной работе мы даем генетическую характеристику клинических штаммов вируса кори, выявленных в 2023–2024 гг., и сравниваем их с глобальными вариантами, а также с российскими штаммами, выявленными в предыдущие годы.
Цель. Молекулярно-генетическая характеристика штаммов вируса кори, выявленных в России в период повышенной заболеваемости в 2023– 2024 гг. в двух крупных городах — Москве и Новосибирске, и их дальнейший филогенетический анализ.
Материалы и методы. В исследование включены 40 полученных в Москве и Новосибирске в 2023–2024 гг. последовательностей генома вируса кори, выделенного из образцов назофарингеального мазка. Данные сравнивали со штаммами, собранными в России в мае 2023 г., депонированными ЦНИИ эпидемиологии Роспотребнадзора, и штаммами, собранными в 2020 и 2021 гг. в Москве.
Результаты. Изученные нуклеотидные последовательности Measles morbillivirus разделены на разные филогенетические группы в пределах генотипа D8. Для образцов генотипа B3, собранных в 2020 и 2023 гг., проведен сравнительный анализ с целью определения региона происхождения. Филогенетический анализ российских и зарубежных вариантов вируса кори позволяет предположить, что штаммы, циркулирующие в настоящее время в России, могут быть разновидностью штаммов, ранее циркулировавших в других странах и независимо распространившихся в России в 2023 г. Проведен анализ наиболее частых нуклеотидных замен в различных генах вируса кори. Определены наиболее вариабельные гены, на основе которых можно расширить филогенетический анализ.
Выводы. Предложенный подход к молекулярно-генетическому исследованию полных и частичных последовательностей геномов клинических штаммов вируса кори, обнаруженных в 2023–2024 гг. в Москве и Новосибирске, дал возможность выделить подгруппы, отличающиеся по происхождению. Сравнение секвенированных нами штаммов Measles morbillivirus с глобальными последовательностями позволило обнаружить близкие последовательности, выявленные как в 2023 г., так и в предыдущие годы на территории различных стран мира. Из анализа эпидемически значимых штаммов Measles morbillivirus следует, что использование гена N позволяет с высокой достоверностью определять основной генотип, однако не является достаточным для изучения путей передачи вируса.
Черняева Е.Н., Морозов K.В., Мацвай A.Д., Гуськова M.С., Некрасов А.Ю., Стеценко И.Ф., Носова А.О., Курская О.Г., Шестопалов А.М., Шипулин Г.A. Молекулярно-генетическая характеристика и филогенетический анализ российских и зарубежных вариантов вируса кори 2020–2024 гг. Экстремальная биомедицина. 2024;26(3):40-51. https://doi.org/10.47183/mes.2024-26-3-40-50
Chernyaeva E.N., Morozov K.V., Matsvay A.D., Guskova M.S., Nekrasov A.Y., Stetsenko I.F., Nosova A.O., Kurskaya O.G., Shestopalov A.M., Shipulin G.A. Molecular and genetic characteristics from phylogenetic analysis of Russian and foreign variants of measles virus 2020–2024. Extreme Medicine. 2024;26(3):40-51. https://doi.org/10.47183/mes.2024-26-3-40-50
Корь — острое инфекционное заболевание с очень высоким уровнем контагиозности, вызываемое РНК-вирусом, принадлежащим к роду Morbillivirus из семейства Paramyxoviridae. Несмотря на существование безопасной и эффективной вакцины, корь остается одной из ведущих причин детской смертности в мире.
Согласно данным ВОЗ, в 2022 г. после многолетнего снижения охвата вакцинацией против кори количество заболевших корью выросло на 18%, а число умерших от инфекции — на 43% (по сравнению с данными на 2021 г., когда количество смертей составило 128 000 человек) [1]. Заболеваемость корью в России по 60 регионам в предыдущие годы колебалась от 0,1 тыс. случаев в 2010 г. до 4,5 тыс. случаев в 2019 г. В 2021 г. был зарегистрирован только один случай, а в 2022 г. — 100 случаев [2]. Во время пандемии SARS-CoV-2 в ряде стран мира было отложено или пропущено введение более 61 млн доз вакцины от кори, в том числе в рамках плановой вакцинации детей, в связи с чем можно ожидать рост заболеваемости уязвимой доли населения и возникновения локальных вспышек кори [3][4].
В начале 2023 г. были выявлены вспышки кори практически во всех регионах мира. К концу ноября 2023 г. в Европейском союзе было зарегистрировано более 42 200 случаев инфицирования вирусом кори, пять из которых закончились смертельным исходом [5][6]. По данным Роспотребнадзора, в 2023 г. в 44 регионах России корью заболело более 13 000 человек (8,9 на 100 тыс. населения). Большая часть случаев (67,9 %) из общего числа зарегистрированных выявлена у детей в возрасте от 0 до 17 лет включительно, два из них закончились смертельным исходом. В 2023 г. в Москве было зарегистрировано 2244 случая кори (17,18 на 100 тыс. населения), а в Новосибирске 266 случаев кори (9,5 на 100 тыс. населения) [7]. Главой Роспотребнадзора отмечено, что эти вспышки носили локальный характер, были быстро купированы и не потребовали введения глобальных ограничительных мер [8].
Кроме локальных вспышек источниками распространения инфекции в России могут быть большие потоки мигрантов и беженцев. По данным ООН на июль 2022 г., в Россию прибыло порядка 1,8 млн украинских беженцев [9]. По данным ВОЗ в Европейском регионе (а именно на Украине) была зарегистрирована самая неблагоприятная ситуация по числу выявленных случаев кори в последние годы (в 2019 г. было зарегистрировано более 57 000 случаев) [10]. В 2023 г. крупные вспышки кори также зафиксированы в странах-соседях России с пониженным миграционным контролем, таких как Азербайджан, Казахстан, Узбекистан, Таджикистан и Армения. В некоторых из них вспышки кори регистрировались и ранее, например в Узбекистане в 2019 г. [11]. Такая обстановка может в ближайшее время привести к значительному росту заболеваемости корью в России, поэтому крайне важно контролировать ситуацию по распространению различных штаммов вируса кори и своевременно проводить филогенетический анализ с целью определения происхождения каждого конкретного штамма.
Для отслеживания и определения происхождения штамма вируса кори используется генотипирование методом секвенирования, чаще всего гена нуклеопротеина, который считается наиболее вариабельным. Известно, что для генотипирования штаммов вируса кори наиболее распространено секвенирование 450 нуклеотидов, кодирующих 150 аминокислот C-концевого домена нуклеопротеина (N450). Только за 2019–2022 гг. в базу данных MeaNS2 было загружено более 11 000 записей о нуклеотидных последовательностях данного участка [12]. Однако последовательности N450 часто бывает недостаточно для разделения эндемичных линий штаммов, циркулирующих в соседних регионах страны. Использование для анализа нуклеотидной последовательности фрагмента генома вируса кори большего размера улучшит разрешение молекулярно-эпидемиологического анализа кори. Наиболее эффективным в данном случае может быть применение полногеномного секвенирования, однако данный метод имеет существенный недостаток в виде высокой цены. Необходимо оценить целесообразность применения расширенного секвенирования фрагментов генома по сравнению с данными полногеномного секвенирования.
Цель работы — молекулярно-генетическая характеристика штаммов вируса кори, обнаруженных в России в период повышенной заболеваемости в 2023–2024 гг. в двух крупных городах — Москве и Новосибирске, и их дальнейший филогенетический анализ.
Во время вспышек заболевания в 2023–2024 гг. у пациентов из Новосибирска и Москвы с диагностированными случаями кори были собраны 40 клинических образцов в Федеральном исследовательском центре фундаментальной и трансляционной медицины (Новосибирск) и Центре стратегического планирования и управления биомедицинскими рисками здоровью ФМБА (ЦСП). Исследованы также архивные образцы штаммов, выявленных в Москве в 2020 г. Для определения штамма вируса кори для всех образцов, включенных в исследование, проведено генотипирование. Для генетического типирования вируса кори используются данные сравнительного анализа нуклеотидных последовательностей С — концевого фрагмента гена нуклеопротеина N (450 нт (нуклеотиды), 1126–1575 нт), для более детальной характеристики у части образцов (n = 15) дополнительно проанализированы фрагменты (850 нт) гена гемагглютинина H. Указанные гены являются наиболее вариабельными структурными генами вируса, в связи с чем лучше всего подойдут для определения происхождения и путей распространения отдельных штаммов вируса кори. Кроме того, для трех образцов вируса кори проведено полногеномное секвенирование. Полученные данные сравнивали со штаммами, собранными в России в мае 2023 г., депонированными в ЦНИИ эпидемиологии Роспотребнадзора (ЦНИИЭ) (полный список образцов вируса кори из России, использованных в работе, приведен в таблице 1).
Для проведения сравнительного филогенетического анализа штаммов вируса кори, распространенных в России в 2020–2024 гг., а также глобальных генетических вариантов использованы как фрагменты генов N и H, так и данные полногеномного секвенирования. В анализ включены образцы вируса кори, полученные от 40 пациентов из Москвы (n = 11) и Новосибирска (n = 29) в 2023–2024 гг., а также два образца из Москвы, выявленные в 2020 и 2021 гг. Все образцы получены с полного информированного согласия доноров и с соблюдением всех необходимых этических норм.
Вирусную РНК выделяли из клинических образцов назофарингеального мазка с помощью ранее разработанного набора для очистки «Рибо-преп» и подвергали обратной транскрипции с помощью «АмплиТест Реверта» («АмплиТест», Россия) [13]. Фрагменты С-терминального участка N-гена, а также фрагмента H-гена амплифицировали с использованием ДНК-полимеразы LongAmp® Taq (New England Biolabs, США) и праймеров, описанных в таблице 2. Секвенирование по Сэнгеру проводили с использованием набора для циклического секвенирования BigDye™ Terminator v3.1 (Thermo Fisher Scientific, США) и генетического анализатора Applied Biosystems 3500 (Thermo Fisher Scientific, США). Три образца секвенировали с помощью полногеномного секвенирования: для приготовления библиотеки использовали тотальную РНК с использованием набора NEBNext® Ultra™ II RNA Library Prep (NEB, США); секвенирование проводилось с применением платформы MiSeq и набора реагентов MiSeq Reagent Kit v2 (500 циклов) (Illumina, США). Праймеры взяты из работы Schulz и др. и далее оптимизированы для нашей задачи [14].
Дополнительно из базы данных GeneBank NCBI получены полногеномные последовательности шести российских образцов вируса кори, изолированных в 2023 г. и исследованных ЦНИИЭ [15]. Кроме того, были отобраны последовательности генов N и H штаммов, обнаруженных в 2019–2024 гг. в различных странах, а также полногеномные последовательности за тот же период из открытой базы данных GenBank. Для более глубокого сравнения в исследование добавлены последовательности геномов вакцинных штаммов, доступные в базе данных GenBank [15].
Метагеномная сборка последовательностей полных геномов вируса кори осуществлялась с использованием программного обеспечения Trimmomatic v0.39 для удаления адаптерных последовательностей и SPAdes genome assembler v3.15.5 для сборки вирусного генома [16]. Последовательности фрагментов генов N и H, полученные секвенированием методом Сэнгера, были объединены в одну для дальнейшего выравнивания и анализа.
Множественное выравнивание было выполнено с использованием пакета MAFFT v7.505 [17]. Филогенетический анализ проводился с использованием программы FastTree версии 2.1.11 SSE3 [18] с использованием модели GTR+CAT (общая обратимая во времени модель с фиксированной скоростью для каждого сайта (аппроксимация модели CAT)) и 100 бутстреп-репликаций. Последовательность генома вируса кори дельфина (AJ608288) использовалась в качестве внешней группы для построения кладограмм.
Анализ филогенетических деревьев проводили с использованием программы FigTree v.1.4.4. Визуализацию результатов сравнительного анализа осуществляли с помощью пакетов Matplotlib 3.6.2 и Seaborn 0.13.2 для языка программирования Python.
Таблица 1. Перечень включенных в исследование образцов, собранных в разных городах России
|
№ п/п |
Внутренний идентификатор образца |
Год взятия образца |
Город обнаружения |
Секвенированный участок генома |
Генотип штамма вируса кори |
Источник данных |
|
1 |
Mea01 |
2023 |
Москва |
WGS |
D8 |
ЦСП ФМБА |
|
2 |
Mea02 |
2023 |
Новосибирск |
WGS |
D8 |
ЦСП ФМБА |
|
3 |
Mea04 |
2020 |
Москва |
WGS |
B3 |
ЦСП ФМБА |
|
4 |
Mea05 |
2023 |
Москва |
N |
D8 |
ЦСП ФМБА |
|
5 |
Mea06 |
2023 |
Новосибирск |
N, H |
D8 |
ЦСП ФМБА |
|
6 |
Mea07 |
2023 |
Новосибирск |
N, H |
D8 |
ЦСП ФМБА |
|
7 |
Mea08 |
2023 |
Новосибирск |
N, H |
D8 |
ЦСП ФМБА |
|
8 |
Mea10 |
2021 |
Москва |
N |
B3 |
ЦСП ФМБА |
|
9 |
Mea11 |
2023 |
Московская область |
N, H |
D8 |
ЦСП ФМБА |
|
10 |
Mea12 |
2023 |
Москва |
N, H |
D8 |
ЦСП ФМБА |
|
11 |
Mea13 |
2023 |
Москва |
N, H |
D8 |
ЦСП ФМБА |
|
12 |
Mea14 |
2023 |
Москва |
N, H |
D8 |
ЦСП ФМБА |
|
13 |
Mea15 |
2023 |
Новосибирск |
N |
D8 |
ЦСП ФМБА |
|
14 |
Mea16 |
2023 |
Новосибирск |
N |
D8 |
ЦСП ФМБА |
|
15 |
Mea17 |
2023 |
Новосибирск |
N |
D8 |
ЦСП ФМБА |
|
16 |
Mea18 |
2023 |
Новосибирск |
N |
D8 |
ЦСП ФМБА |
|
17 |
Mea20 |
2023 |
Новосибирск |
N |
D8 |
ЦСП ФМБА |
|
18 |
Mea21 |
2023 |
Новосибирск |
N |
D8 |
ЦСП ФМБА |
|
19 |
Mea22 |
2023 |
Новосибирск |
N |
D8 |
ЦСП ФМБА |
|
20 |
Mea23 |
2023 |
Новосибирск |
N |
D8 |
ЦСП ФМБА |
|
21 |
Mea24 |
2023 |
Новосибирск |
N |
D8 |
ЦСП ФМБА |
|
22 |
Mea25 |
2023 |
Новосибирск |
N |
D8 |
ЦСП ФМБА |
|
23 |
Mea26 |
2023 |
Новосибирск |
N |
D8 |
ЦСП ФМБА |
|
24 |
Mea27 |
2023 |
Новосибирск |
N |
D8 |
ЦСП ФМБА |
|
25 |
Mea28 |
2023 |
Новосибирск |
N |
D8 |
ЦСП ФМБА |
|
26 |
Mea29 |
2023 |
Новосибирск |
N |
D8 |
ЦСП ФМБА |
|
27 |
Mea30 |
2023 |
Новосибирск |
N |
D8 |
ЦСП ФМБА |
|
28 |
Mea31 |
2023 |
Новосибирск |
N |
D8 |
ЦСП ФМБА |
|
29 |
Mea32 |
2023 |
Новосибирск |
N |
D8 |
ЦСП ФМБА |
|
30 |
Mea33 |
2023 |
Новосибирск |
N |
D8 |
ЦСП ФМБА |
|
31 |
Mea34 |
2023 |
Новосибирск |
N |
D8 |
ЦСП ФМБА |
|
32 |
Mea35 |
2023 |
Новосибирск |
N |
D8 |
ЦСП ФМБА |
|
33 |
Mea36 |
2023 |
Новосибирск |
N |
D8 |
ЦСП ФМБА |
|
34 |
Mea37 |
2023 |
Новосибирск |
N |
D8 |
ЦСП ФМБА |
|
35 |
Mea39 |
2023 |
Москва |
N, H |
D8 |
ЦСП ФМБА |
|
36 |
Mea43 |
2023 |
Москва |
N, H |
D8 |
ЦСП ФМБА |
|
37 |
Mea44 |
2023 |
Москва |
N, H |
D8 |
ЦСП ФМБА |
|
38 |
Mea45 |
2023 |
Москва |
N, H |
D8 |
ЦСП ФМБА |
|
39 |
Mea46 |
2023 |
Москва |
N, H |
D8 |
ЦСП ФМБА |
|
40 |
Mea47 |
2024 |
Новосибирск |
N, H |
D8 |
ЦСП ФМБА |
|
41 |
Mea48 |
2024 |
Новосибирск |
N, H |
B3 |
ЦСП ФМБА |
|
42 |
Mea49 |
2024 |
Новосибирск |
N, H |
D8 |
ЦСП ФМБА |
|
43 |
OR290097 |
2023 |
Нет данных |
Полный геном |
D8 |
ЦНИИЭ |
|
44 |
OR290098 |
2023 |
Нет данных |
Полный геном |
B3 |
ЦНИИЭ |
|
45 |
OR290099 |
2023 |
Нет данных |
Полный геном |
D8 |
ЦНИИЭ |
|
46 |
OR290100 |
2023 |
Нет данных |
Полный геном |
D8 |
ЦНИИЭ |
|
47 |
OR290101 |
2023 |
Нет данных |
Полный геном |
D8 |
ЦНИИЭ |
|
48 |
OR290102 |
2023 |
Нет данных |
Полный геном |
D8 |
ЦНИИЭ |
Таблица подготовлена авторами по собственным данным
Таблица 2. Список последовательностей праймеров для секвенирования генов N и H
|
Секвенируемый ген |
Нуклеотидная последовательность прямого праймера |
Нуклеотидная последовательность обратного праймера |
|
ген N |
TGGAGCTATGCCATGGGAGT |
TAACAATGATGGAGGGTAGG |
|
ген H (регион1) |
CTGAAATTGTCTCYGGCTTC |
GACCCAGGATTGCATGTC |
|
ген H (регион 2) |
GATTTCAGCAACTGYATGGT |
CTGATGTCTGRGTGACATCATG |
Таблица подготовлена авторами по собственным данным
В результате генотипического анализа установлено, что большая часть образцов (n = 44), собранных в 2023–2024 гг., относятся к генотипу D8 и только один — к генетическому варианту B3 (табл. 1). Филогенетический анализ частичного секвенирования гена N (рис. 1) показал, что в пределах генотипа D8 большинство образцов из России сформировали кластер, в который также вошли и некоторые штаммы, обнаруженные за рубежом:
В составе данного филогенетического кластера можно обозначить два субкластера, один из которых сформирован образцами из Новосибирска (А), в состав второго вошли три образца из Москвы и два из Новосибирска (Б). Штаммы, секвенированные ЦНИИЭ, сформировали филогенетическую группу с образцами Mea2 и Mea43 из Москвы.
Только две последовательности гена N образцов вируса кори, выявленных в 2023 г. в России, удалось классифицировать как представителей филогенетической группы B3: Mea48, зафиксированный в Новосибирске, и OR290098, секвенированный ЦНИИЭ, с неизвестным регионом сбора (рис. 1, кластер В). Данные образцы не сформировали кластер с последовательностями генов вирусных штаммов, выявленных в России в 2020 и 2021 гг. Помимо российских образцов, еще 3 штамма из Бразилии, США и Ирана, обнаруженные в 2023 г., и два штамма из Китая и Швейцарии, выявленные в 2024 г., попали в филогенетический кластер B3. Большинство штаммов Measles morbillivirus, отнесенных к генотипу B3, были выявлены до 2023 г.
Согласно филогенетическому анализу фрагментов генов N и H, российские образцы сформировали субкластер внутри генотипа D8. Результаты позволили уточнить филогенетическую структуру популяции исследованных российских штаммов. В частности, не подтвердился субкластер Б внутри генетической группы D8, сформированный образцами Mea7, Mea8, Mea13, Mea44 и Mea45 (рис. 2, 3) на основании анализа только гена N.
Сравнение результатов филогенетического анализа с использованием только фрагмента N и последовательности, полученной при объединении последовательностей фрагментов генов N и H, позволяет сделать вывод о том, что секвенирование гена N является достаточным для определения генетической группы штаммов вируса кори, но для изучения путей передачи вируса необходимо проводить расширенный анализ с участием дополнительных участков генома. Дополнительное исследование гена Н даст возможность точнее определить генетическое сходство между образцами вируса кори.
Секвенирование полных геномов проведено для двух образцов генотипа D8, относящихся к двум филогенетически отдаленным и выявленным в различных городах штаммам: Mea1 (Москва, 2023 г.) и Mea2 (Новосибирск, 2023 г.). Полногеномное секвенирование проведено также для образца Mea4, полученного в 2020 г. и относящегося к генотипу B3. Сравнительный анализ геномов выявил 145 нуклеотидных различий между Mea1 и Mea2, из которых 39 попадают в межгенное пространство, а 106 замен — в различные кодирующие белок участки генов. Наибольшее их число приходится на гены L (52 замены), N (17 замен) и H (15 замен). Полученный результат позволяет сделать предположение, что данные штаммы вируса кори могут иметь различное происхождение.
Для филогенетического анализа полногеномных последовательностей в исследование были включены все доступные геномы из базы данных GenBank, выявленные за последние 5 лет и относящиеся к группам B3, D8, а также последовательности вакцинных штаммов. Филогенетический анализ полногеномных последовательностей российских штаммов и последовательностей, обнаруженных в других регионах мира, показал, что российские штаммы наиболее близки с образцами, выделенными в США и Индии в 2019 г., а также Японии и Новой Зеландии в 2023 г. Соответствующие данные представлены на рисунке 4.
Различия в образованных подгруппах внутри генотипов вируса кори, полученные в результате филогенетического анализа штаммов вируса, заставляют задуматься об эффективности проведения анализа на основе только гена N. Для дальнейшего исследования данного вопроса был проведен сравнительный анализ филогенетических деревьев, сформированных на основе гена N, генов N и H, а также генами полногеномных последовательностей, путем оценки дистанции Хэмминга между парами штаммов внутри и между генотипами B3 и D8. Результаты анализа (рис. 5) отражают дискриминирующую способность филогенетического анализа, основанного на различном количестве генов.
Результаты анализа наглядно демонстрируют достаточную дискриминирующую способность анализа фрагмента гена N при определении генотипов D8 и B3, однако недостаточную эффективность разделения филогенетических групп внутри генотипов. Одновременный анализ генов N и H позволяет с более высокой точностью разделять штаммы как внутри филогенетических групп, так и между ними, повышая статистическую значимость и достоверность исследования. Анализ всех кодирующих белок областей и полногеномных последовательностей значительно повышает точность исследования путей передачи вирусов внутри одной филогенетической группы (рис. 5).
Для оценки вариабельности генов Measles morbillivirus проводили сравнительный анализ последовательностей генов в исследуемой выборке, состоящей из полноразмерных геномов штаммов вируса кори из различных стран, относящихся к генетическим группам D8 и B3. Сравнительный анализ позволил определить общее количество однонуклеотидных вариантов в генах белков N, L, M, F, PVC и H. Оценивали количество и долю синонимичных и несинонимичных нуклеотидных вариантов (рис. 6). Наибольшее число нуклеотидных замен обнаружено в гене L, который является самым большим и кодирует белок вирусной РНК полимеразы размером 2183 аминокислоты. Профиль распределения мутаций в гене М, кодирующем матричный белок вируса размером 335 аминокислот, оказался наиболее отличающимся от остальных генов. В частности, данный ген имел больше несинонимичных замен (n = 114), чем синонимичных (n = 71). В составе гена PVC, который кодирует несколько вирусных белков (фосфопротеин (P), неструктурный белок V и белок С), также обнаружено больше несинонимичных замен (n = 88), чем синонимичных (n = 69).
Анализ частот нуклеотидных замен в генах Measles morbillivirus (рис. 6) демонстрирует, что ген N не является наиболее вариабельным, а гены M и H являются важными мишенями для уточнения филогенетических связей между штаммами патогена.
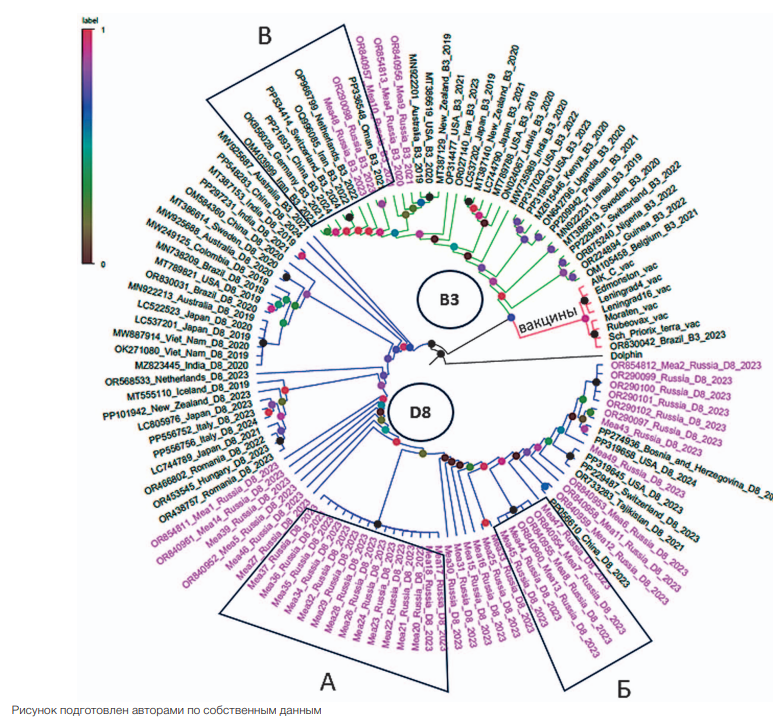
Рисунок подготовлен авторами по собственным данным
Рис. 1. Филогенетическая связь штаммов Measles morbillivirus на основании данных секвенирования фрагмента гена N
Синим цветом обозначена филогенетическая группа D8, зеленым — филогенетическая группа B3, красным — группа вакцинных штаммов. Розовым текстом выделены геномы из России, включенные в анализ, секвенированные в 2020, 2021 и 2023–2024 гг. Субкластер А сформирован образцами из Новосибирска; субкластер Б — образцами из Москвы и Новосибирска, относящимися к генотипу D8. Субкластер В филогенетической группы В3 включает два образца — обнаруженный в 2023 г. в Новосибирске Mea48 и OR290098, секвенированный ЦНИИЭ, с неизвестным регионом сбора.
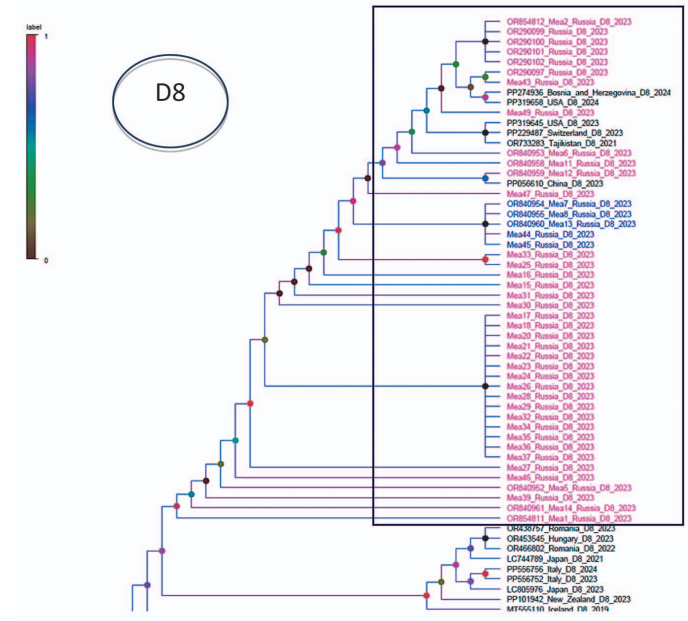
Рисунок подготовлен авторами по собственным данным
Рис. 2. Положение исследованных российских образцов в кластере D8, полученное в результате филогенетического анализа, проведенного с использованием фрагмента последовательности гена N. Кластер, сформированный исследованными российскими образцами, выделен прямоугольной рамкой, образцы окрашены в синий и фиолетовый цвет теста. Образцы Mea7, Mea8, Mea13, Mea44 и Mea45, образующие субкластер Б, выделены синим шрифтом
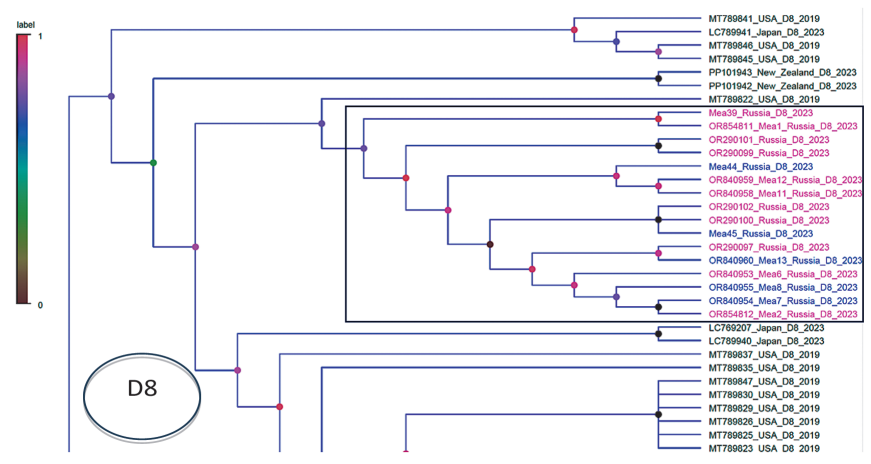
Рисунок подготовлен авторами по собственным данным
Рис. 3. Положение исследованных российских образцов в кластере D8, полученное в результате филогенетического анализа, проведенного с использованием объединенной последовательности генов N и H. Кластер, сформированный исследованными российскими образцами, выделен прямоугольной рамкой, образцы окрашены в синий и фиолетовый цвет теста. Образцы Mea7, Mea8, Mea13, Mea44 и Mea45, образующие субкластер Б, выделены синим шрифтом
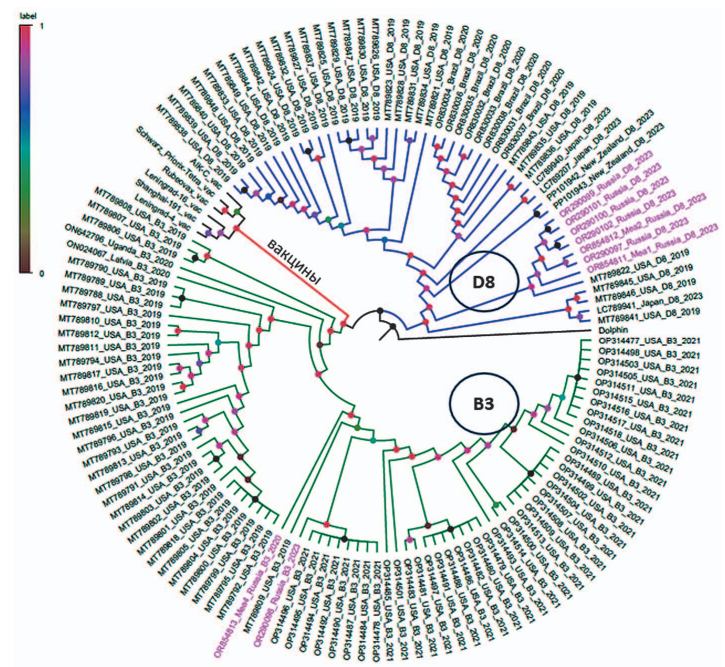
Рисунок подготовлен авторами по собственным данным
Рис. 4. Филогенетическая связь полногеномных последовательностей штаммов вируса кори, обнаруженных в России и других странах. Синим цветом обозначена филогенетическая группа генотипа D8, зеленым — группа генотипа B3, красным — группа вакцинных штаммов. Розовым текстом выделены геномы из России, включенные в анализ, секвенированные в 2020 и 2023 гг.
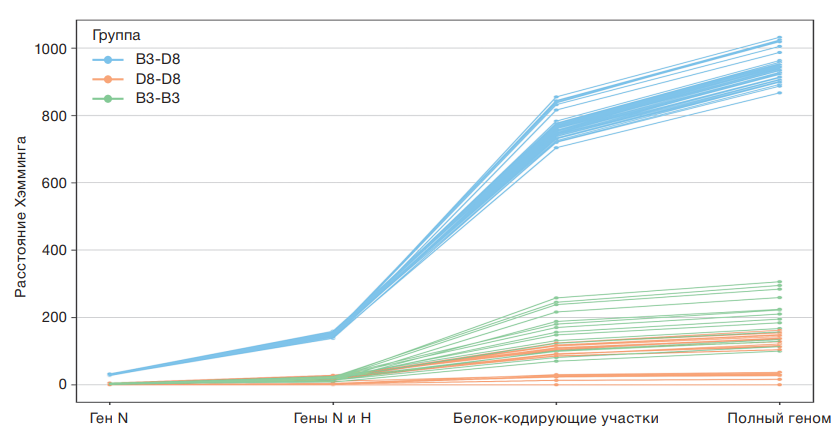
Рисунок подготовлен авторами по собственным данным
Рис. 5. Попарное расстояние Хэмминга между последовательностями штаммов вируса кори, относящихся к генетическим группам D8 и B3. По вертикали показаны значения расстояния Хэмминга при попарном сравнении последовательностей, по горизонтали отражены исследуемые регионы генома вируса кори — фрагмент гена N, объединение фрагментов генов N и H, объединение всех кодирующих белок последовательностей, а также полный геном. Цветом показаны значения для попарного сравнения последовательностей внутри групп B3 и D8, а также между группами штаммов: синий — B3–D8, зеленый — D8–D8, оранжевый B3–B3
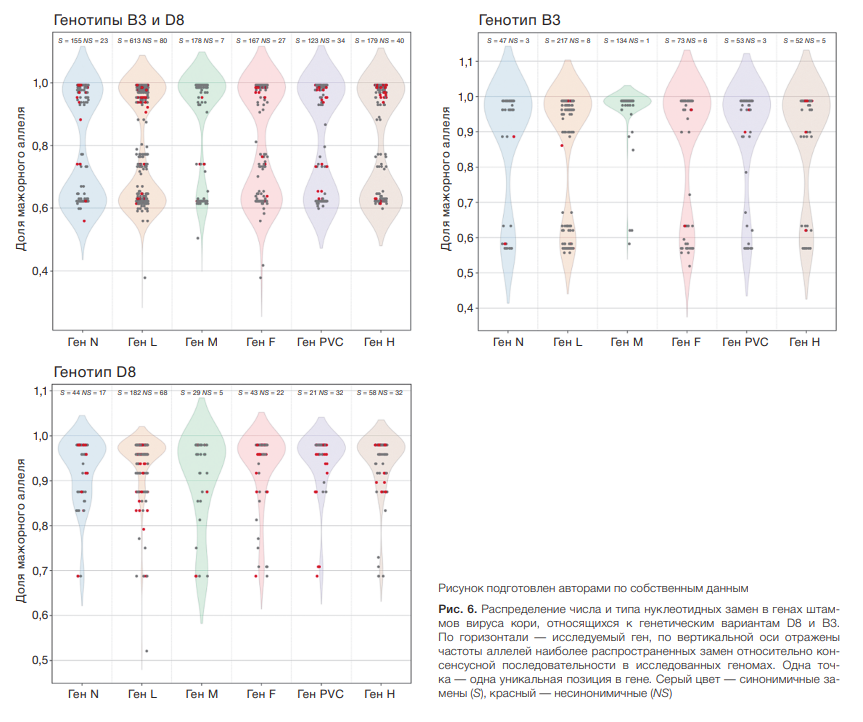
Рисунок подготовлен авторами по собственным данным
Рис. 6. Распределение числа и типа нуклеотидных замен в генах штаммов вируса кори, относящихся к генетическим вариантам D8 и B3.
По горизонтали — исследуемый ген, по вертикальной оси отражены частоты аллелей наиболее распространенных замен относительно консенсусной последовательности в исследованных геномах. Одна точка — одна уникальная позиция в гене. Серый цвет — синонимичные замены (S), красный — несинонимичные (NS)
Результаты сравнительного анализа исследованных нами образцов вируса кори говорят о генетической схожести российских циркулирующих вариантов со штаммами, которые были обнаружены в других регионах мира, например Центральной Европе и Азии. Опубликованные за 2019–2022 гг. данные согласуются с полученными нами результатами. Так, генотипы D8 и B3 являются доминирующими в глобальной картине генетического разнообразия вируса кори. Однако показано, что их соотношение варьируется в различных регионах мира. В частности, генотип B3 доминировал в Африканском, Американском, Восточно-Средиземноморском регионах, а генотип D8 являлся доминирующим в Европейском, Северо-Восточноазиатском и Западном Тихоокеанском регионах [19]. Информация, представленная на сайте базы данных Nextstrain [20], также свидетельствует о доминирующем распространении генотипов B3 и D8 в различных регионах мира с 2020 г. (рис. 7).
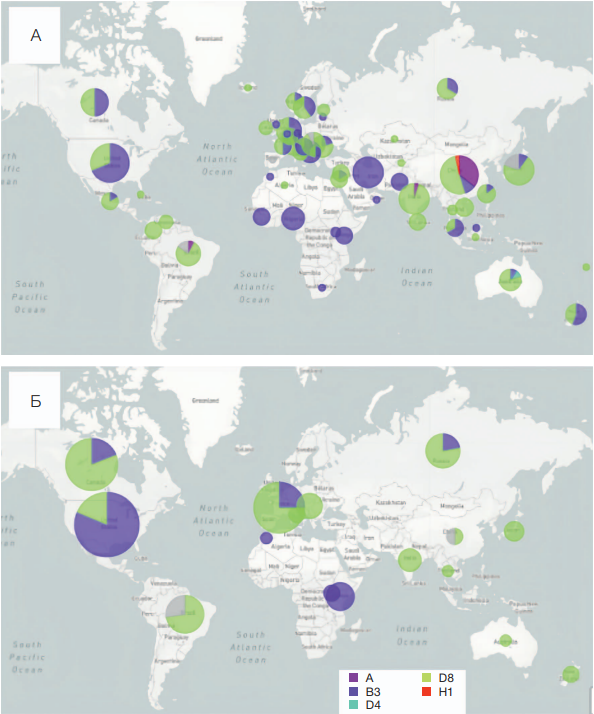
Рисунок подготовлен авторами
Рис. 7. Глобальное распространение известных генотипов вируса кори. Инфографика, построенная на основании: A — данных секвенирования фрагмента гена N, Б — данных секвенирования полных геномов в период с января 2020 по сентябрь 2024 г. Рисунок сформирован с использованием интернет-ресурса https://nextstrain.org/1/
Большинство исследованных вариантов, выявленных нами в 2023–2024 гг., относятся к генотипу D8, в который входит широко распространенная группа штаммов [12][19]. Всего в открытой базе данных GenBank содержится более 6 тысяч последовательностей генов или геномов штаммов, относящихся к данной группе.
Среди российских штаммов, помимо генотипа D8, обнаружены также штаммы, относящиеся к генотипу B3. Вариант генотипа B3 был идентифицирован только в двух российских образцах в 2023 г., однако оба образца, которые были получены в Москве в 2020 и 2021 гг., относятся к группе, распространенной в данный момент в странах Африки, Европы и Северной Америки. Среди геномов или фрагментов геномов Measles morbillivirus, полученных из образцов пациентов в 2023 г. и доступных в открытых базах данных, таких как база Национального центра биотехнологической информации (NCBI), лишь 60 относились к данной группе и встречались преимущественно в США и Иране. Из филогенетического анализа этой группы штаммов видно, что варианты, обнаруженные в России в 2020 и 2023 гг., не образуют общий кластер, из чего можно предположить их различное происхождение. В работе Ерохова и др. [12] показано, что во вспышках 2018–2020 гг. имели место генетические линии и варианты вируса кори, не циркулирующие на территории России; они, по всей вероятности, были импортированы из соседних стран. К аналогичному выводу пришли Кузьменков и др. [21], когда при проведении ретроспективного анализа вспышек кори в Астраханской области выделили процесс миграции вариантов вируса из соседних стран в ряд ключевых факторов образования новых вспышек.
Исходя из описанного выше, можно предположить, что большинство вспышек кори в России в 2023–2024 гг. вызваны именно вариантами группы D8, которые также обнаружены в различных регионах мира в этот период. Это подтверждается статистическими данными о распределении генотипов среди случаев заражения корью за период с 2020 по 2024 г. в Европейском регионе [6].
Большую часть изученных нуклеотидных последовательностей Measles morbillivirus, связанных с последними вспышками кори в России, можно разделить на филогенетические подгруппы по месту происхождения или пути распространения в пределах генотипа D8. Из зависимости расстояния между подгруппами (рис. 5) видно, что разрешающая способность филогенетического анализа обусловлена величиной фрагмента используемой в нем нуклеотидной последовательности генома вируса. Сравнительный анализ структур филогенетических деревьев, полученных при изучении последовательности гена N, а также комбинации генов N и H, показал, что использование только гена N с высокой достоверностью определяет основной генотип, но не позволяет исследовать пути передачи вируса. Сравнение изменчивости генов Measles morbillivirus также показало, что ген N не является наиболее вариабельным, а для исследования путей передачи вируса требуется изучение дополнительных генов, таких как М и H. В дальнейшем для достижения оптимального соотношения трудозатрат и разрешающей способности анализа путей передачи вируса кори следует использовать как минимум три наиболее вариабельных гена для выбранного генотипа, например N, H и M.
Предложенный подход к молекулярно-генетическому исследованию полных и частичных последовательностей геномов клинических штаммов вируса кори, обнаруженных в 2023–2024 гг. в Москве и Новосибирске, сделал возможным выделить подгруппы, отличающиеся по происхождению. Среди секвенированных штаммов, выявленных в 2023 г., зафиксировано также два образца, относящихся к генотипу B3 и не образующих общую по происхождению группу с последовательностями того же генотипа, найденными в Москве в 2020–2021 гг.
Сравнение секвенированных нами штаммов Measles morbillivirus с глобальными последовательностями позволило обнаружить близкие последовательности, обнаруженные как в 2023 г., так и в предыдущие годы на территории различных стран мира.
Исследование гена N при анализе эпидемически значимых штаммов Measles morbillivirus позволяет с высокой достоверностью определять основной генотип, однако не является достаточным для изучения путей передачи вируса. Рекомендуется использовать и другие наиболее вариабельные гены, такие как М и Н.
1. World Health Organization. Measles. [Онлайн]; Доступно: https://www.who.int/news-room/fact-sheets/detail/1#:~:text=1%20vaccination%20averted%2056%20million,the%20age%20of%205%20years [Accessed: September 5, 2023]
2. Российский статистический ежегодник, 2023. Федеральная служба государственной статистики; 2023. [Онлайн]; Доступно: https://rosstat.gov.ru/storage/mediabank/Ejegodnik_2023.pdf [Accessed: July 5, 2024]
3. Global Measles Outbreaks. Centers for Disease Control and Prevention. [Онлайн]; Доступно: https://www.cdc.gov/globalhealth/1/data/global-1-outbreaks.html [Accessed: July 5, 2024]
4. Dixon MG, Ferrari M, Antoni S. Li X, Portnoy A, Lambert B, et al. Progress towards regional meats elimination – worldwide, 2000-2020. Morb Mortal Wkly Rep. 2021;70(45):1563. https://doi.org/10.15585/mmwr.mm7045a1
5. Bedford H., Elliman D., Measles rates are rising again, BMJ 2024;384:q259. https://doi.org/10.1136/bmj.q259
6. World Health Organization. Measles. [Онлайн]; Available: https://www.who.int/europe/ru/publications/m/item/1-and-rubella-monthly-update-who-european-region-june-2024 [Accessed: July 5, 2024]
7. Заболеваемость корью в России в 2023 году оказалась рекордной за 30 лет. [Онлайн]; Доступно: https://medvestnik.ru/content/news/Zabolevaemost-koru-v-Rossii-v-2023-godu-okazalas-rekordnoi-za-30-let.html [Accessed: July 5, 2024]
8. RIA News. [Онлайн]; Доступно: https://ria.ru/20230420/kor-1866513301.html [Accessed: July 5, 2024]
9. RIA News. [online]; Доступно: https://ria.ru/20230420/kor-1866513301.html [Accessed: July 5, 2024] (In Russ.).
10. Refugees from Ukraine recorded by country. [Online]; Доступно: https://data.unhcr.org/en/situations/ukraine. [Accessed: July 5, 2024]
11. The World Health Organization. 1 – number of reported cases. [Online]; Available: www.who.int/data/gho/data/indicators/indicator-details/GHO/1--number-of-reported-cases. [Accessed: July 15, 2023]
12. Bryantseva E, Matnazarova G, Tursunov D and Saidkasimova N. Epidemiological features of 1 infection during an outbreak in Tashkent city. EBWFF 2023 – International Scientific Conference Ecological and Biological Well-Being of Flora and Fauna (Part 1). 2023;V.420.
13. https://doi.org/10.1051/e3sconf/202342005014
14. Ерохов ДВ, Жердева ПЕ, Рубальская ТС, Фролов РА, Тихонова НТ. Глобальное генетическое разнообразие вирусов кори, краснухи, паротита в 2019–2022 гг. Современные проблемы эпидемиологии, микробиологии и гигиены. 2022;108-111.
15. Носова АО, Богословская ЕВ, Шипулин ГА. Современные подходы и перспективы развития лабораторной диагностики кори. Клиническая микробиология и антимикробная химиотерапия. 2023;25(1).
16. Schulz H, Hiebert J, Frost J, McLachlan E, Severini A. Optimisation of methodology for whole genome sequencing of 1 Virus directly from patient specimens. Journal of Virological Methods. 2022;299:114348.
17. https://doi.org/10.1016/j.jviromet.2021.114348
18. Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, et al. Nucleic Acids Res. 2013;41(Database issue):D36-42.
19. https://doi.org/10.1093/nar/gks1195.
20. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. Journal of Computational Biology. 2012;19(5):455–77.
21. https://doi.org/10.1089/cmb.2012.0021
22. Katoh K, Standley DM. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Molecular Biology and Evolution. 2013;30(4):772–80. https://doi.org/10.1093/molbev/mst010
23. Price MN, Dehal PS, Arkin AP (2010) FastTree 2 – Approximately Maximum-Likelihood Trees for Large Alignments. Plos one 5(3):e9490.
24. https://doi.org/10.1371/journal.pone.0009490
25. Чехляева ТС, Ерохов ДВ, Жердева ПЕ, Тихонова НТ. Генотипы вируса кори в российской федерации и их разнообразие в 2018–2020 гг. Эпидемиологический надзор за актуальными инфекциями: новые угрозы и вызовы. 2021;327–9.
26. T.S.Chekhlyaeva, D.V. Erokhov, P.E. Zherdeva, N.T. Tikhonova. Measles virus genotypes in the Russian Federation and their diversity in 2018-2020. Epidemiologic surveillance of current infections: new threats and challenges. 2021;327-9. 2021;327–9 (In Russ.).
27. https://doi.org/10.21145/978-5-6046124-2-2_2021https://doi.org/10.21145/978-5-6046124-2-2_2021
28. Nextstrain. Real-time tracking of pathogen evolution [cited 2024 Aug. 12]; [Online]; Доступно: https://nextstrain.org/1/ [Accessed: July 5, 2024]
29. Кузменков МВ, Спиренкова АЕ, Ахмерова РР, Рвачев ВС. Международный научно-исследовательский журнал. — 2024;3(141).
30. M.V. Kuzmenkov, A.E. Spirenkova, R.R. Akhmerova, V.S. Rvachev. Current epidemiologic features of measles on the territory of the Astrakhan region in 2013-2023. International Research Journal.2024;141(3) (In Russ.).
31. https://doi.org/10.23670/IRJ.2024.141.68
Черняева Eкатерина Николаевна, канд. биол. наук
Москва
Морозов Кирилл Владимирович
Москва
Мацвай Aлина Дмитриевна, канд. биол. наук
Москва
Гуськова Mария Сергеевна, канд. физ.-мат. наук
Москва
Некрасов Андрей Юрьевич
Москва
Стеценко Иван Федорович
Москва
Носова Анастасия Олеговна
Москва
Курская Ольга Григорьевна, канд. мед. наук
Новосибирск
Шестопалов Александр Михайлович, докт. биол.
Новосибирск
Шипулин Герман Александрович, канд. мед. наук
Москва
Черняева Е.Н., Морозов K.В., Мацвай A.Д., Гуськова M.С., Некрасов А.Ю., Стеценко И.Ф., Носова А.О., Курская О.Г., Шестопалов А.М., Шипулин Г.A. Молекулярно-генетическая характеристика и филогенетический анализ российских и зарубежных вариантов вируса кори 2020–2024 гг. Экстремальная биомедицина. 2024;26(3):40-51. https://doi.org/10.47183/mes.2024-26-3-40-50
Chernyaeva E.N., Morozov K.V., Matsvay A.D., Guskova M.S., Nekrasov A.Y., Stetsenko I.F., Nosova A.O., Kurskaya O.G., Shestopalov A.M., Shipulin G.A. Molecular and genetic characteristics from phylogenetic analysis of Russian and foreign variants of measles virus 2020–2024. Extreme Medicine. 2024;26(3):40-51. https://doi.org/10.47183/mes.2024-26-3-40-50
119121, Москва, Погодинская ул., д. 10, стр. 1
Тел.: +7 (495) 540-61-71, доб.: 4190, 4191, 4192
E-mail: extrememedicine@cspfmba.ru












