



Contents
Scroll to:
https://doi.org/10.47183/mes.2024-242
Scroll to:
Introduction. Nanopore sequencing technologies have become routine methods in science and medicine, being widely used in the study of pathogen diversity and distribution and playing a key role in field epidemiology.
Objective. Comparative evaluation of the functional capabilities of third-generation MinION and Nanoporus sequencers in the detection of pathogens in biological material, including comparison of the as-determined taxonomic composition with the results obtained using the second-generation MiSeq (Illumina) reference platform.
Materials and methods. A total of 138 archival DNA samples with known taxonomic composition (14 families, 20 genus, and 43 species of viral and bacterial pathogens; altogether 169 pathogens) were analyzed. MinION and Nanoporus nanopore sequencers with original R9.4.1 and R10.4.1 flow cells (ONT), as well as the high-performance MiSeq (Illumina) platform were used for preliminary identification of the composition of samples containing different titers of pathogen nucleic acids belonging to various taxonomic groups. Comparative evaluation of the obtained data (number of sequences, average read quality scores (Qscore) for each nucleotide, GC-content of sequences, sequence length distribution, read duplication level) was performed using the MultiQC bioinformatics tool (version 1.20).
Results. The MinION and Nanoporus devices identified 98.8% and 97.6% of pathogens, respectively, including understudied or new viruses. The use of the latest-version flow cell on both devices significantly reduced the share of low-quality reads. The findings demonstrate a high degree of correlation between the results obtained by the second- and third-generation sequencers, which confirms the comparability and interchangeability of these technologies for the purposes of pathogen nucleic acid identification.
Conclusions. The study results demonstrate the potential of MinION and Nanoporus nanopore sequencers for epidemiologic surveillance. These devices are capable of identifying pathogens of different nature with high accuracy and, due to their compactness and portability, facilitating the diagnostics and monitoring of infectious diseases.
Grigoryan D.A., Stetsenko I.F., Gukov B.S., Matsvay A.D., Shipulin G.A. Comparative evaluation of MinION and Nanoporus nanopore sequencers in identification of pathogen nucleic acids. Extreme Medicine. 2025;27(1):64-73. https://doi.org/10.47183/mes.2024-242
Sequencing technologies have become routine methods in various areas of molecular biology, due to their capacity to promptly and reliably detect mutations and genetic variations in viruses, identify new pathogens, predict their evolutionary changes, track the dynamics of distribution in populations, and analyze phylogenetic relationships [1–3]. The method of nanopore sequencing was first introduced by Oxford Nanopore Technologies (ONT) in 2014 with the MinION device [4]. This technology possesses a number of unique advantages, which determine its indispensability in modern medicine and science [5]. The ability to sequence long DNA and RNA fragments has made it possible to detect structural variations and epigenetic modifications [6–8]. However, the key advantage of this sequencing method consists in the compactness of nanopore sequencers, their ability to operate by connecting to a laptop USB interface, and low requirements for laboratory equipment. This renders such devices applicable in various conditions, including field studies [9].
Nanopore sequencing technology has already made a substantial impact on various fields of medicine, including diagnosis and treatment of genetic diseases [10][11], personalized medicine [2][12], and cancer research [6][13][14]. Due to its speed and mobility, nanopore sequencing is a convenient tool for epidemiologic surveillance and outbreak control [9][15–17]. In particular, during the COVID-19 pandemic, nanopore sequencing played an essential role in rapid identification of virus strains and detection of new genetic variations of pathogens [12][17][18]. In addition, this technology has proven to be a reliable alternative to routine methods of sequencing complete virus genomes [15][19]. These properties are particularly important in the context of global pandemics and outbreaks of new infections, since the promptness of data collection is crucial for the decision-making process.
The Russian market, along with the original third-generation platforms by Oxford Nanopore Technologies, also offers a similarly functional domestic device referred to as Nanoporus. This device is designed for nanopore sequencing using original flow cells by Oxford Nanopore Technologies.
In this work, we carry out a comparative evaluation of the functional capabilities of third-generation MinION and Nanopore sequencers in detecting pathogens in biological material, including comparison of the taxonomic composition identified with their use and the results obtained using the second-generation MiSeq (Illumina) reference platform.
During two stages of the study, 138 samples of archival DNA from a laboratory collection with known taxonomic composition and different titers of pathogen nucleic acid belonging to different taxonomic groups were analyzed. These included 14 families, 20 genus, and 43 species of pathogens of a viral and bacterial nature, totaling 169 pathogens. Identification of the pathogenic composition of the studied material had been previously performed by high-throughput sequencing on the MiSeq platform (Illumina). Based on the data of local alignment of nucleotide and protein sequences, we analyzed similarity indices with sequences from the database used for taxonomic identification.
The following reagent sets were used during the preparation of amplicon DNA libraries. End Repair of double-stranded DNA fragments and matrix-free adenylation were performed using a NEBNext Ultra II End Repair/dA-Tailing Module reagent kit (New England Biolabs). Adaptor sequence ligation from PCR Barcoding Expansion 1-96 (EXP-PBC096) (ONT) was performed using a Blunt/TA Ligase Master Mix reagent (New England Biolabs). Barcoding of libraries was performed using PCR Barcoding Expansion 1-96 (EXP-PBC096) (ONT). All kits were used according to the manufacturer’s instructions.
DNA libraries for sequencing on a R9.4.1 flow cell were prepared using a Ligation Sequencing Kit (SQK-LSK109) (ONT) and then loaded into the flow cell. DNA libraries for sequencing on a R10.4.1 flow cell were prepared using a Ligation Sequencing Kit V14 (SQK-NBD114) reagent kit (ONT) followed by loading into the flow cell. The sequence of sequencing reagents was determined taking technical and methodological considerations into account. At the primary stage of the study, when using the R9.4.1 flow cell for the first time, sequencing was initially performed on the MinION sequencer to ensure a predictably stable operation of the cell. This order was set to minimize potential risks associated with a possible decrease in the stability or functionality of the cell after its use on the Nanoporus sequencer. At the second step, in order to test the Nanoporus sequencer in terms of its effects on cell functionality for subsequent use in the MinION, sequencing was performed initially on the Nanoporus and then on the MinION.
The detected signal from the devices was recorded using the MinKNOW software version 23.11.4; the basecalling of data in pod5 format was performed by the Dorado software version 7.2.13 (ONT). In order to ensure the correctness of sequencing quality comparison and taxonomic identification by the tested devices, the equal number of reads were selected for each sample representing a random sample generated by the SeqKit bioinformatics tool (version v2.8.0). The quality of reads in fastq format was assessed by using Trimmomatic bioinformatic tools (version 0.32), FastQC (version v0.12.0). Comparative evaluation of the data obtained (number of sequences, average read quality scores (Qscore) for each nucleotide, GC-content of sequences (guanine-cytosine content), sequence length distribution, read duplication level) was performed using the MultiQC bioinformatic tool (version 1.20). Taxonomic identification of the viral composition of the samples was performed using the PathogenID software (FMBA, Russia). As a reference tool for comparing the results of determining the taxonomic composition of samples, the data obtained on the second-generation MiSeq platform (Illumina) were used.
For comparative evaluation of the tested sequencers, the investigated infectious agents were divided into five groups according to their taxonomic identification index (% identity) obtained by high-throughput sequencing. The following identifiers were assigned to virus groups: Group 1 — 30 pathogens, 100–97% identity, Group 2 — 29 pathogens, 96–94% identity, Group 3 — 39 pathogens, 93–90% identity, Group 4 — 35 pathogens, 89–80% identity, Group 5 — 24 pathogens, 79–71% identity. The presented pathogen groups can be used to simulate the analysis of divergent pathogen groups, new strains, species in particular.
Using the MinKNOW software, the stability of data transmission from the device to the control computer was assessed. No significant fluctuations and failures in signal transmission, as well as deviations from the uniform distribution of the DNA translocation rate through the pore were registered during the analysis of the graphs obtained from the MinION and Nanoporus devices using the R10.4.1 flow cell (Fig. 1; A2, B2). The graphs of temperature maintenance throughout the entire sequencing process indicated no disturbances in the operation of the sequencers; temperature fluctuations were insignificant and remained within the permissible limits. The corresponding information is presented in Fig. 1; A1, B1.
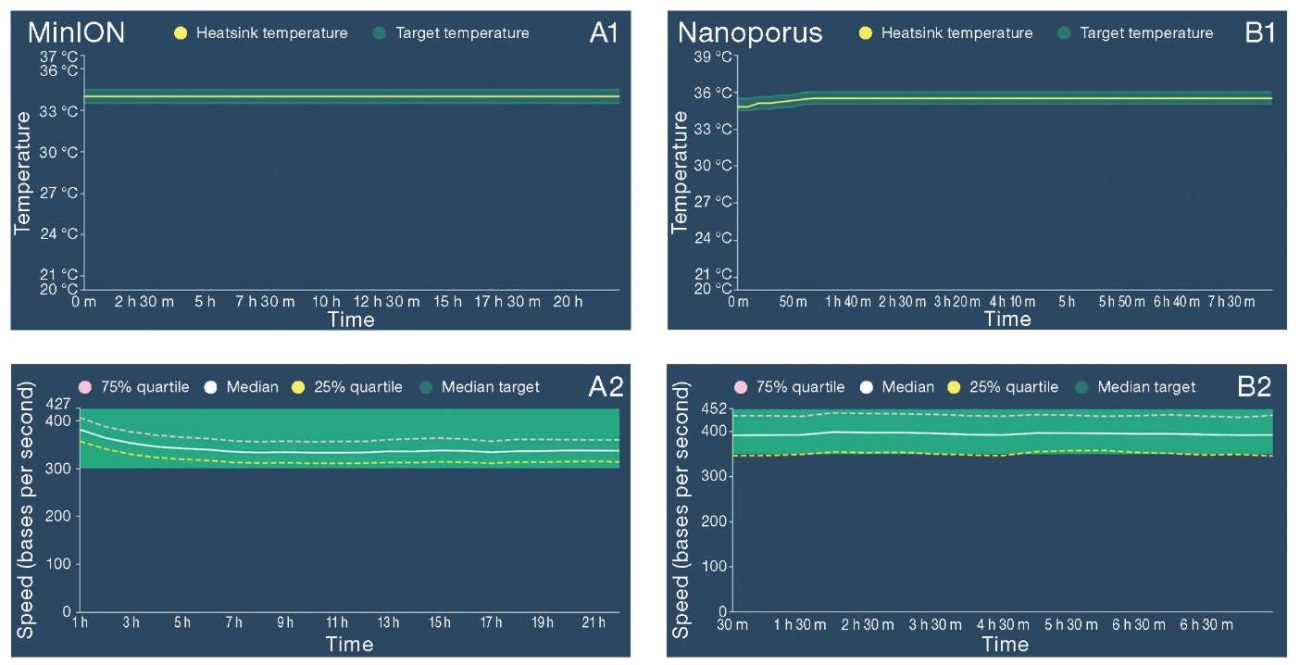
Figure prepared by the authors
Fig. 1. Graphical representation of key parameters of the MinION and Nanoporus sequencers
Note: A1, B1 — maintaining the temperature regime during sequencing; A2, B2 — the rate of DNA translocation through the pore.
The graphs were obtained using the MinKNOW software, version 23.11.4.
The quality parameters of reads obtained from both old- and new-type flow cells for MinION and Nanoporus sequencers were compared (Fig. 2). An analysis of the presented graphs revealed an increase in the Qscore parameter, which reflects the accuracy of base identification in the reads, and a decrease in the level of duplications when using a flow cell of the latest R10.4.1 version. All the presented metrics for assessing the quality of reads obtained from the two compared nanopore sequencers were also found to be consistent.
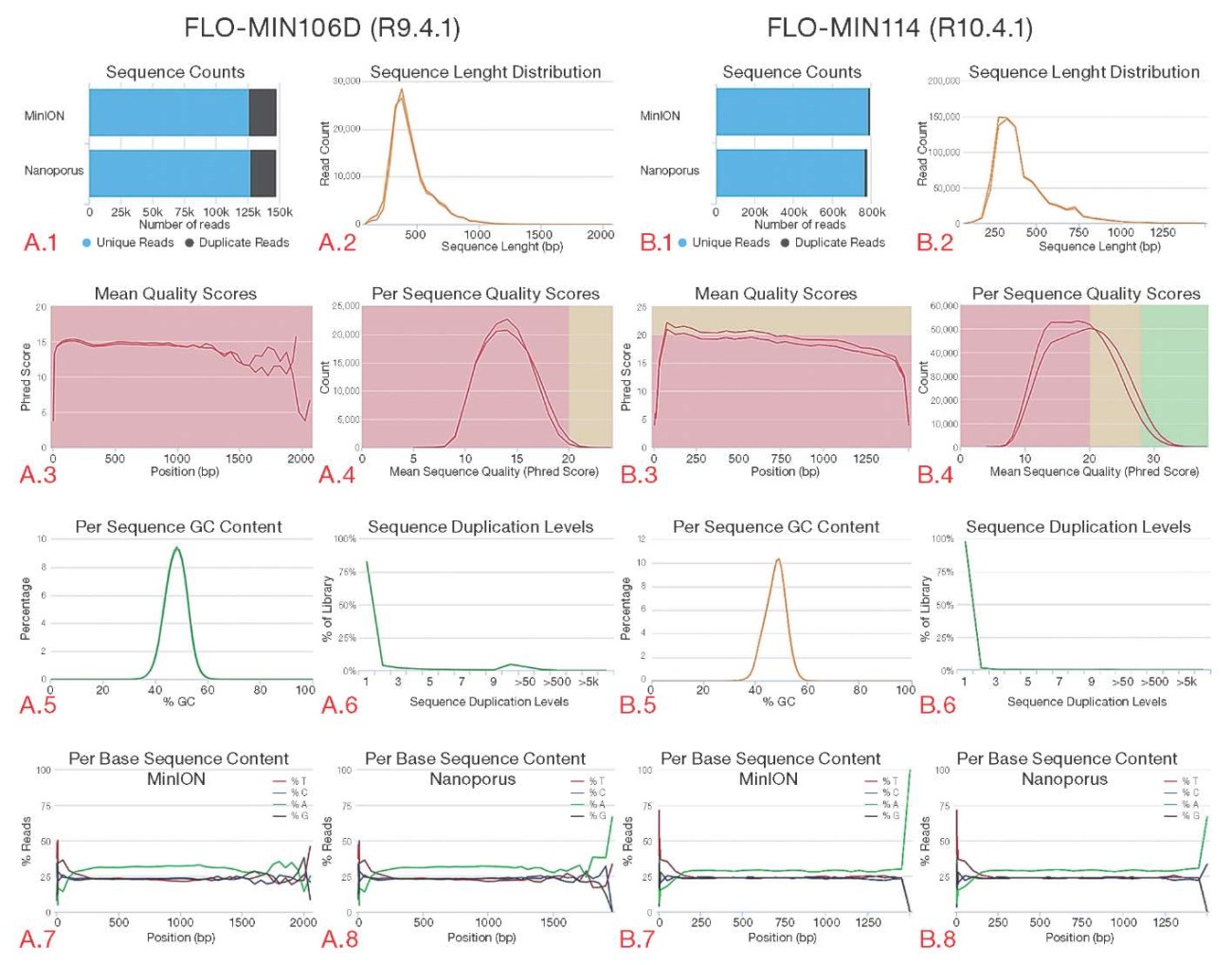
Figure prepared by the authors
Fig. 2. Comparison of quality indicators of reads obtained from MinION and Nanoporus sequencers
Note: graph A.1 — total number of sequences obtained from each device; graph A.2 — distribution of reads by read length; graph A.3 — average read quality values (Qscore) for each nucleotide; graph A.4 — number of reads depending on the quality indicator (Qscore); graph A.5 — GC content in sequences, %; graph A.6 — level of read duplications; graph A.7 — nucleotide composition of reads obtained by the MinION sequencer; graph A.8 — nucleotide composition of reads obtained by the Nanoporus sequencer.
The corresponding data for the R10.4.1 flow cell is shown in graphs B.1–B.8.
The graphs correspond to data recorded using R9.4.1 and R10.4.1 flow cells.
Read quality scores were obtained by the MultiQC bioinformatics tool (version 1.20).
Additional indicators of sequencing quality, namely metrics N50, N95, and N5, as well as the percentage of reads with quality above Q20 and Q30 were calculated; the corresponding data are presented in Table 1. A comparative evaluation of sequence percentage with a quality of above Q20 and Q30 found that when using the earlier version of the flow cell (R9.4.1), about 70% of the data demonstrated a quality below Q20 and about 97.3% had a quality below Q30 on both sequencers. At the same time, when the R10.4.1 flow cell of the latest version was used, the proportion of data with a quality below Q20 was less than 55% and below Q30 was less than 76%. There were no significant differences in N50, N95, N5, and the percentage of sequences with a quality above Q20 and Q30 between the two different third-generation devices.
Table 1. Sequencing quality indicators on the original R10.4.1 and R9.4.1 flow cells on the third-generation sequencers: Nanoporus and MinION
|
Quality assessment parameters |
Flow cell R10.4.1, MinION sequencer |
Flow Cell R10.4.1, Nanoporus sequencer |
Flow cell R9.4.1, MinION sequencer |
Flow cell R9.4.1, Nanoporus sequencer |
|
Average read length, units of nucleotides |
407.8 |
417.1 |
447 |
435 |
|
Maximum read length, units of nucleotides |
1500 |
1500 |
1399 |
1388 |
|
N50 |
411 |
425 |
454 |
446 |
|
N5 |
250 |
256 |
289 |
275 |
|
N95 |
900 |
904 |
871 |
860 |
Table prepared by the authors using their own data
The studied material contained fragments of pathogen genomes of the following families:
Pseudomonadaceae, Circoviridae, Adenoviridae, Coronaviridae, Orthomyxoviridae, Parvoviridae, Polyomaviridae, Astroviridae, Caliciviridae, Picornaviridae, Solemoviridae, Hepeviridae, Partitiviridae, Tymoviridae.
The Adenoviridae family included 12 specimens and 5 pathogen species with viral loads ranging from 7.22% to 0.33% of reads per specimen according to high-throughput sequencing data. As a result of processing the data obtained by MinION and Nanoporus devices, the taxonomic composition was confirmed in 11 (92%) and 10 (83%) samples, respectively. The Circoviridae family counted 35 specimens and 11 different pathogen species with viral loads ranging from 57.27% to 0.04% of reads based on Illumina sequencing data. Data from MinION and Nanoporus sequencers resulted in confirmed taxonomic composition in 34 (97%) and 33 (94%) samples, respectively. Although identification of the target pathogen by nanopore sequencers was not performed for 100% of samples containing viruses of these families (Adenoviridae and Circoviridae), a more detailed virus typing by nanopore data was obtained during alignment of nucleotide sequences to reference databases for a number of samples.
According to the high-throughput sequencing data obtained on the Illumina platform, the Coronaviridae family counted 70 samples and 9 different pathogen species with viral loads ranging from 49.52% to 0.15% of reads. The Orthomyxoviridae family sample counted 6 samples and 1 species with viral loads ranging from 28.97% to 1.32% of reads. The selection of Pseudomonadaceae samples counted 12 samples and 1 species. The test group of the Parvoviridae family counted 15 samples and 11 species of pathogens with viral loads ranging from 28.12% to 0.22% of reads. The Picornaviridae family counted 4 samples and 4 species of pathogens with viral loads ranging from 11.53% to 0.3% of reads. While the Astroviridae, Caliciviridae, Polyomaviridae, Solemoviridae, Tymoviridae, Partitiviridae, and Hepeviridae families counted single specimens, identification of the taxonomic composition of the Coronaviridae, Parvoviridae, Picornaviridae, and Orthomyxoviridae families was carried out in 100% of specimens during third-generation sequencing on MinION and Nanoporus devices. A total of 167 (98.8%) and 165 (97.6%) pathogens were identified by the MinION and Nanoporus nanopore sequencers, respectively.
For the most represented virus families, the percentage of reads attributable to the target infectious agent was compared. For a more accessible visualization of the comparison, data were taken on a logarithmic scale and analyzed for three pairs of sequencers: MiSeq and MinION (A), MiSeq and Nanoporus (B), and MinION and Nanoporus (C) (Fig. 3). The plots showed an increase in correlation between sequencing results along with an increase in viral load in the samples, particularly between Nanopore sequencing data at load levels more than two logarithmic units. At low viral loads, deviations were found, particularly between Illumina and third-generation sequencing data.
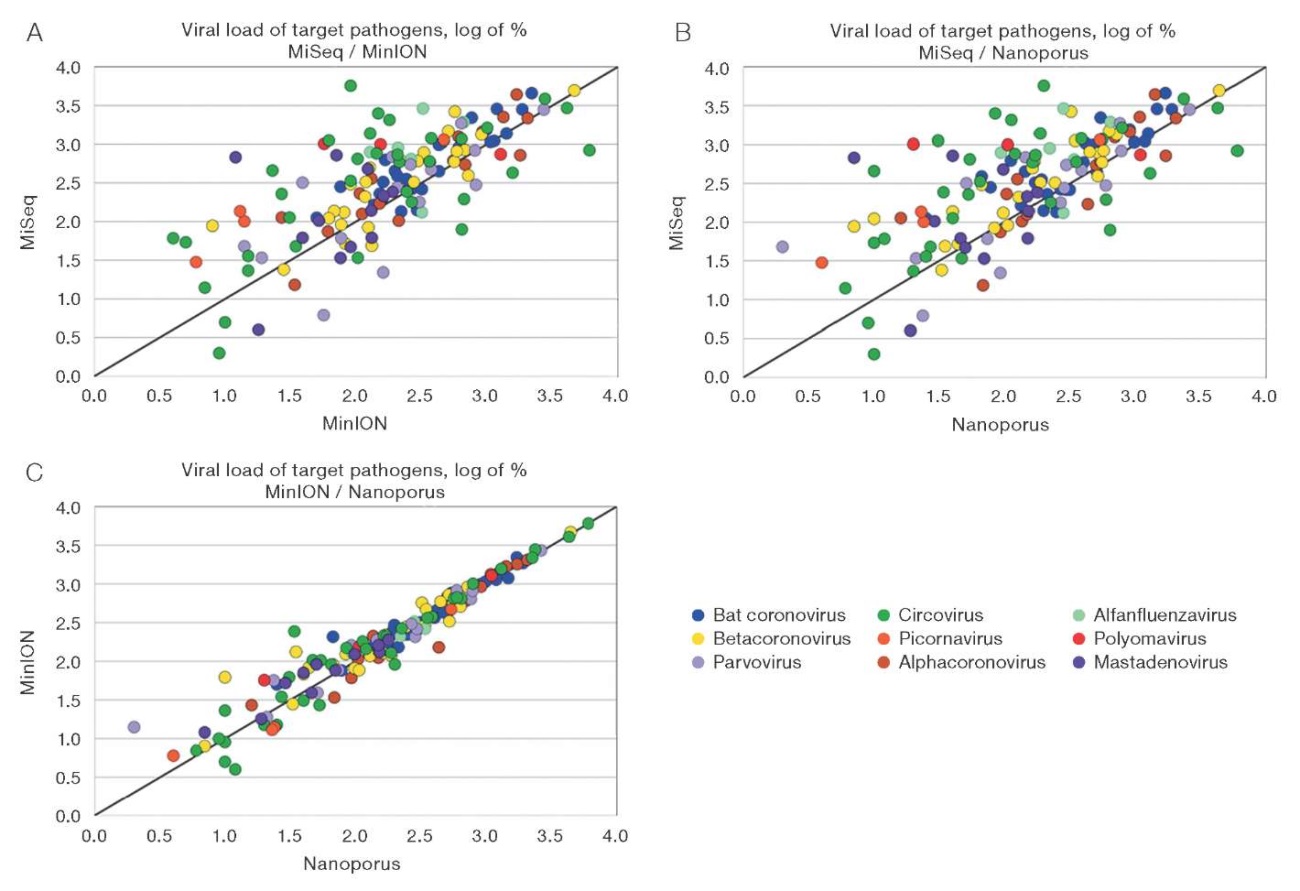
Figure prepared by the authors
Fig. 3. Comparison of the percentage of reads of target pathogens of the most represented families
Note: graph A — MiSeq and MinION sequencers; graph B — MiSeq and Nanoporus sequencers; graph C — MinION and Nanoporus sequencers.
Point on the graphs — one virus under study.
Viral load values are presented on a logarithmic scale.
The Pearson correlation coefficient was calculated to assess the relationship between the percent viral load values on the three devices. An analysis of data collected from both flow cells showed the following results. The correlation coefficient between MiSeq and MinION platforms was r = 0.567 (p ≤ 0.05), indicating a moderate positive relationship. Between MiSeq and Nanoporus, a coefficient of r = 0.544 (p ≤ 0.05) was recorded, also indicating a moderate positive relationship. The highest correlation value was observed between Nanoporus and MinION sequencers, equal to r = 0.993 (p ≤ 0.05), indicating an almost complete correspondence of the results between the two devices.
In the course of the study, groups of infectious agents formed according to the range of taxonomic identity (% identity) were compared according to the average values of taxonomic identification indicators (e-value, % identity, alignment length (nucleotide base pairs), percentage of reads of the target virus) obtained on the tested devices (Fig. 4).
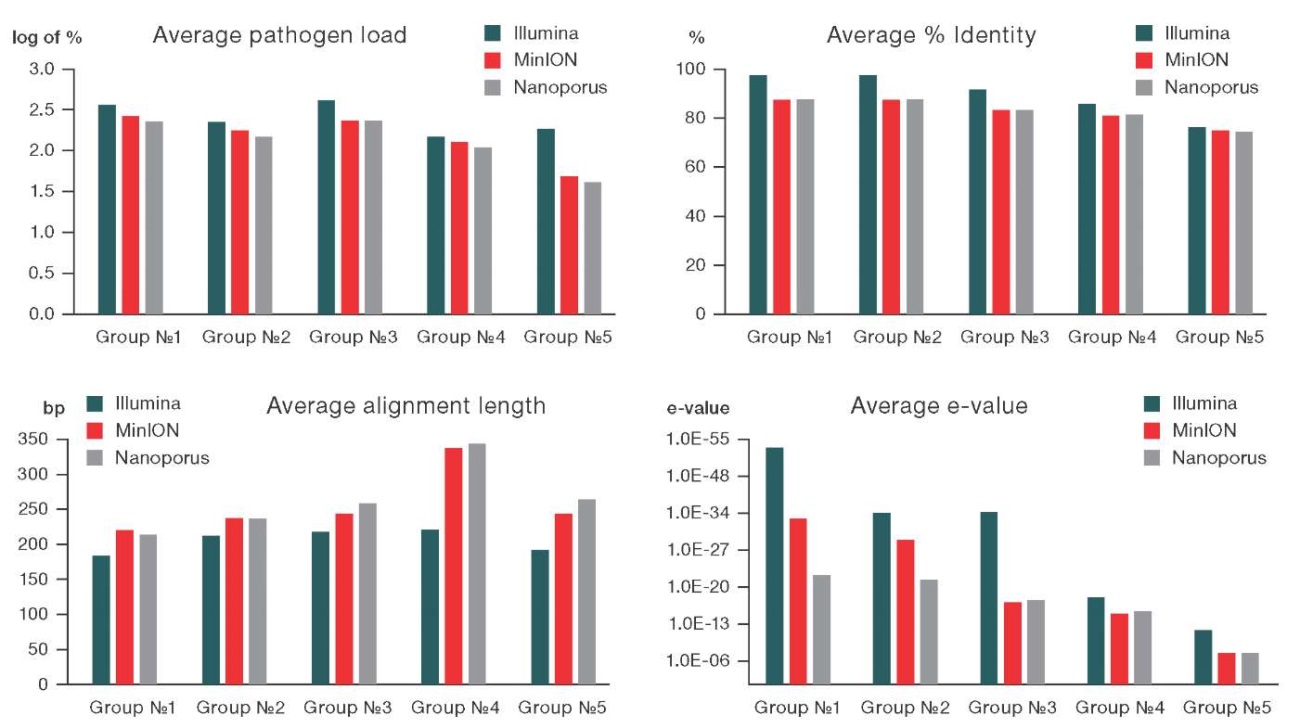
Figure prepared by the authors
Fig. 4. Comparison of average values of taxonomic identification parameters (pathogen load, % identity, alignment length, e-value) obtained as a result of sequencing on MiSeq, MinION, and Nanoporus sequencers
Note: the analysis was performed based on data obtained from flow cells of the latest two versions.
For all presented pathogen groups, the % identity and e-value values obtained on the MiSeq platform were slightly higher than those obtained on nanopore sequencers. At the same time, the index of sequence alignment length showed an inverse relationship. The histograms indicated a high degree of concordance in the detection of infectious agents between the three devices.
To visualize the differences in the reliability of taxonomic identification provided by the tested sequencers, we simulated the variation distribution of viral load and percent identity based on the data obtained from the three devices (Fig. 5). Based on the results of the first stage of the study (95 samples) (A), we observed the following distribution pattern: on the MiSeq platform, the main data cluster was in the range of high identity (90–100%) and medium viral load (2-3 logarithmic units); when using the R9.4.1 flow cell, the MinION platform showed a dense cluster of data in the area of high identity (more than 90%) and a relatively high viral load (2–3 logarithmic units); data from the Nanoporus sequencer were distributed more broadly along the identity axis, however, the main dispersion cluster was also within 90% identity.
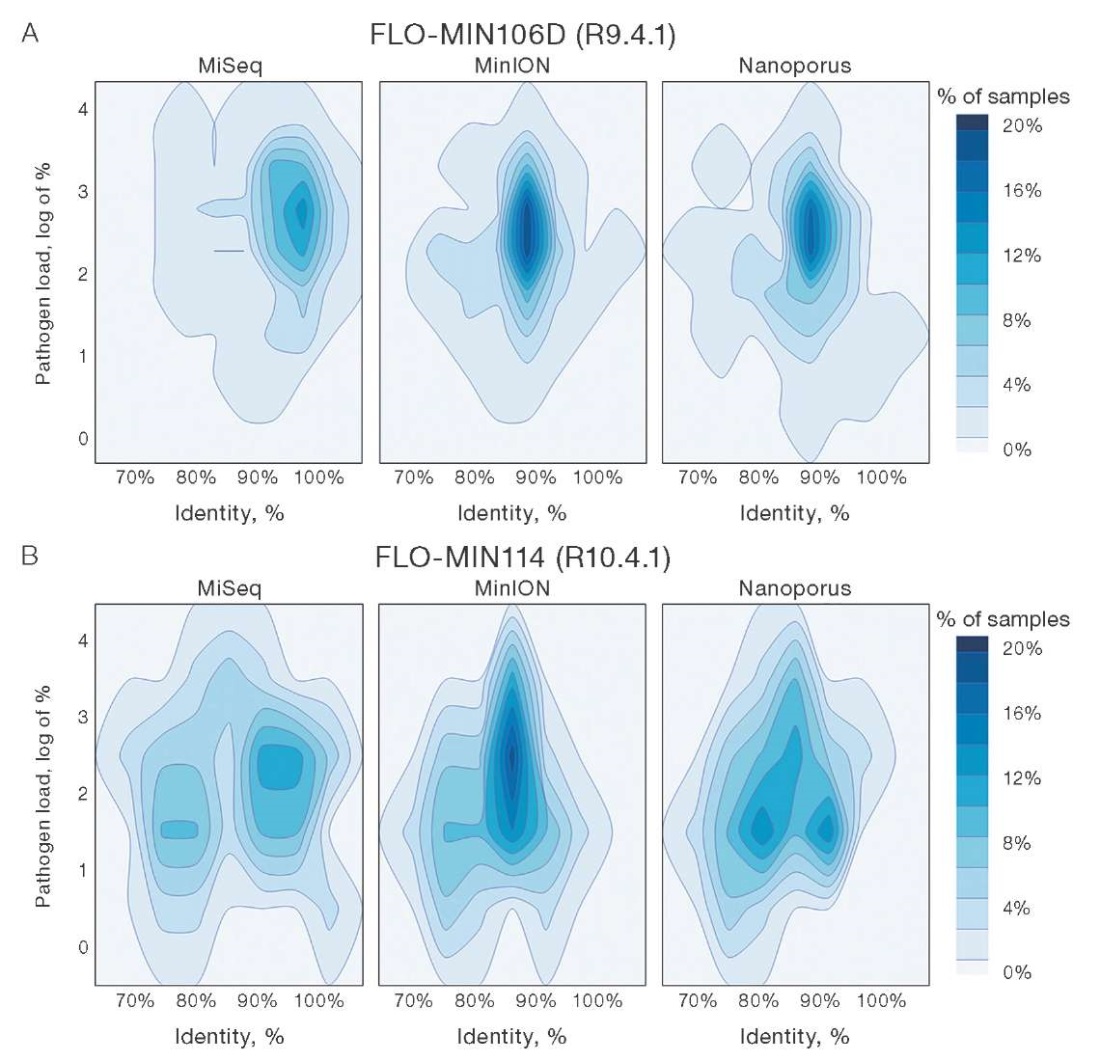
Figure prepared by the authors
Fig. 5. Dispersion distribution of pathogen load and percent identity for data from MiSeq, MinION, and Nanoporus sequencers
Note: graphs correspond to data recorded using R9.4.1(A) and R10.4.1(B) flow cells.
The results of the second stage of the study (43 samples suspected to contain nucleic acids of poorly studied pathogens) (B) showed the following data profile. The MiSeq platform revealed a shift in data density towards lower identity and viral load, which is explained by the peculiarities of the tested material. When using the R10.4.1 flow cell, data from the MinION were concentrated in a narrow range of 85% identity and a relatively high viral load (2–3 logarithmic units). The Nanoporus sequencer showed a less dense data distribution relative to its counterpart, although demonstrating a comparable viral load.
For a long period of time, Oxford Nanopore Technologies (ONT) has been the only developer offering nanopore sequencing solutions. However, over the past year, several similar platforms have entered the market. These include, in particular, the Chinese QNome-3841 and QNome-3841hex sequencers by QitanTech, which have already found application in forensic genetics and full bacterial genome studies [27][28]; CycloneSEQ by MGI, which has showed significant results in de novo genome assembly, as well as in metagenomic and single-cell sequencing according to a study published by the platform developers [20]. At the time of this research, a comparative evaluation of CycloneSEQ and Gnome with such systems as MinION was not possible due to the recent announcement of the platforms. The Nanoporus Nanopore Sequencer, which is an analog of the well-known MinION device, was announced in late 2023 and commercialized in early 2024. At the time of preparing this article, we were unable to find any publicly available studies devoted to a direct comparison of the characteristics of these devices; therefore, our conclusions are based purely on our own experimental data.
During testing of the Nanopore sequencer, no violations or abnormalities in operation were detected. The compatibility of the device with the original flow cells of the two latest versions, library preparation kits and software by Oxford Nanopore Technologies was confirmed. The data on the rate of DNA translocation through nanopores did not reveal any signal distortions, indicating that there were no significant deviations from the expected indicators of the quality of information transfer to the control computer. The indicators of temperature maintenance stability by nanopore sequencers showed minimal temperature fluctuations, which is important for stable operation of nanopores, ensuring sequencing accuracy, preventing library degradation, and maintaining optimal conditions for the functioning of enzymes involved in the sequencing process. In the framework of this analysis, the equipment was found to meet the stated characteristics and functional requirements.
An updated R10.4.1 flow cell significantly improves sequencing accuracy and stability, as evidenced by the increase in average Qscore values on both nanopore platforms compared to the previous flow cell version. The existing studies in the field confirm that the latest version of ONT flow cells provides significant improvement in accuracy and quality of reads [21]. According to the data provided by the authors, the percentage of base pairs with Q15 read quality for the R10.4.1 flow cell is six times higher than that for the R9.4.1 version. In the case of bacterial genome assembly, the authors were able to assemble 97% of the genome through the earlier version of the flow cell, and this value increased to 98% in the case of R10.4.1. Our study shows a similar improvement: the proportion of reads with quality below Q20 and Q30 decreases significantly; the proportion of low-quality reads below Q20 decreases from 70% to 55% and below Q30 from 97.3% to 76%. This change indicates an improvement in sequencing accuracy and reliability when using ONT’s new V14 chemistry and R10.4.1 flow cells.
Second-generation sequencing is recognized as a highly accurate and reliable method for detecting nucleic acids of infectious agents. Due to its high quality of nucleotide reads, this method can be classified as a leading technology among alternative molecular diagnostic methods [22]. However, nanopore sequencing technology (ONT) offers a number of unique advantages, such as compactness and mobility, thus standing out against the complex optical systems required for second-generation sequencing. These characteristics of ONT allow efficient use of the technology in resource-limited settings and rapid field studies, which is an important factor in the introduction of this method into epidemiologic surveillance processes.
Previous studies demonstrated the potential of ONT as an alternative to high-throughput second-generation platforms. In particular, in a study evaluating the identification of pathogens of bacterial nature using MinION and Illumina sequencers (the type of second-generation devices is not specified), both devices were used to successfully identify species, serotypes, MLST profiles, and subtypes of Shiga-toxin in Escherichia coli (STEC) isolates [23]. Another study on the identification of the bacterial composition of a reference sample containing eight different pathogens found that classification performance at the family and genus level prevailed when using MinION. However, at the species level, pathogen identification was found to be more accurate on the MiSeq platform. It is important to note that the R9.4.1 flow cell version was used for MinION [24]. A number of other studies evaluating the efficiency of using different generations of sequencing for the detection of viral pathogens, in particular, representatives of the genera Alphavirus [25] and Adenovirus [26], also demonstrate only a slight advantage of second-generation platforms in terms of identification quality compared to the results obtained on nanopore sequencers. Our data support the conclusions of the above studies by demonstrating a high correlation between pathogen detection results of different generations of sequencers. The MinION and Nanoporus devices identified 98.8% and 97.6% of pathogens, respectively, of the infectious agents detected by the MiSeq platform. It was also found that an increase in pathogen load in the sample led to an increase in the level of similarity between second- and third-generation sequencing results. A particularly high concordance is noted between the data obtained by MinION and Nanoporus platforms as the average value of pathogen nucleic acid titer in the tested samples is exceeded. In this regard, MinION and Nanoporus can be considered interchangeable in the context of pathogen identification and quantification tasks.
Our study evaluated the ability of nanopore sequencers to detect novel or poorly understood viral variations. The control material under study contained samples for which preliminary identification of pathogen composition using MiSeq platform showed low alignment quality parameters. This may indicate limited representation of these pathogen variations in existing databases or complete absence of information thereon. The analysis of variance distribution and quality parameters for taxonomic identification demonstrated significant alignment between second- and third-generation data. This confirms the capability of MinION and Nanoporus nanopore sequencers to identify previously unknown or poorly studied viruses at a level comparable to high-performance platforms.
The Nanopore device demonstrated compatibility with the latest two versions of flow cells, library preparation kits, and software by Oxford Nanopore Technologies. This allows its integration into existing laboratory processes without the need for significant modifications to working protocols. The results of the conducted comparative evaluation confirmed a high level of consistency of pathogen taxonomic identification data obtained using third-generation MinION and Nanopore sequencers with the results provided by the second-generation MiSeq platform. Based on the data obtained, no limitations for the use of MinION and Nanoporus nanopore sequencers in laboratory pathogen detection studies have been identified.
1. Brown BL, Watson M, Minot SS, Rivera MC, Franklin RB. MinIONTM nanopore sequencing of environmental metagenomes: a synthetic approach. GigaScience. 2017;6(3):gix007. https://doi.org/10.1093/gigascience/gix007
2. Schmidt K, Mwaigwisya S, Crossman LC, Doumith M, Munroe D, Pires C. Identification of bacterial pathogens and antimicrobial resistance directly from clinical urines by nanopore-based metagenomic sequencing. J Antimicrob Chemother. 2017;72(1):104–14. https://doi.org/10.1093/jac/dkw397
3. Ciuffreda L, Rodríguez-Pérez H, Flores C. Nanopore sequencing and its application to the study of microbial communities. Comput Struct Biotechnol J. 2021;19:1497–511. https://doi.org/10.1016/j.csbj.2021.02.020
4. Jain M, Olsen HE, Paten B, Akeson M. The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol. 2016;17(1):239. https://doi.org/10.1186/s13059-016-1103-0
5. Leggett RM, Clark MD. A world of opportunities with nanopore sequencing. J Exp Bot. 28 2017;68(20):5419–29. https://doi.org/10.1093/jxb/erx289
6. Ahmed YW, Alemu BA, Bekele SA, Gizaw ST, Zerihun MF, Wabalo EK. Epigenetic tumor heterogeneity in the era of single-cell profiling with nanopore sequencing. Clin Epigenetics.2022;14(1):107. https://doi.org/10.1186/s13148-022-01323-6
7. Searle B, Müller M, Carell T, Kellett A. Third-Generation Sequencing of Epigenetic DNA. Angew Chem. 2023;135(14):e202215704. https://doi.org/10.1002/ange.202215704
8. Parker MT, Knop K, Sherwood AV, Schurch NJ, Mackinnon K, Gould PD. Nanopore direct RNA sequencing maps the complexity of Arabidopsis mRNA processing and m6A modification. Wan Y, Hardtke CS. eLife. 2020;9:e49658. https://doi.org/10.7554/eLife.49658
9. Quick J, Loman NJ, Duraffour S, Simpson JT, Severi E, Cowley L. Real-time, portable genome sequencing for Ebola surveillance. Nature. 2016;530(7589):228–32. https://doi.org/10.1038/nature16996
10. Ameur A, Kloosterman WP, Hestand MS. Single-Molecule Sequencing: Towards Clinical Applications. Trends Biotechnol. 2019;37(1):72–85. https://doi.org/10.1016/j.tibtech.2018.07.013
11. Sun X, Song L, Yang W, Zhang L, Liu M, Li X. Nanopore Sequencing and Its Clinical Applications. Methods Mol Biol Clifton NJ. 2020;2204:13–32. https://doi.org/10.1007/978-1-0716-0904-0_2
12. Wang M, Fu A, Hu B, Tong Y, Liu R, Liu Z. Nanopore Targeted Sequencing for the Accurate and Comprehensive Detection of SARS-CoV-2 and Other Respiratory Viruses. Small. 2020;16(32):2002169. https://doi.org/10.1002/smll.202002169
13. Chen Z, He X. Application of third-generation sequencing in cancer research. Med Rev. 2021;1:000010151520210013. https://doi.org/10.1515/mr-2021-0013
14. Lau BT, Almeda A, Schauer M, McNamara M, Bai X, Meng Q. Single-molecule methylation profiles of cell-free DNA in cancer with nanopore sequencing. Genome Med. 2023;15(1):33. https://doi.org/10.1186/s13073-023-01178-3
15. Faizuloev E, Mintaev R, Petrusha O, Marova A, Smirnova D, Ammour Y. New approach of genetic characterization of group A rotaviruses by the nanopore sequencing method. J Virol Methods. 2021;292:114114. https://doi.org/10.1016/j.jviromet.2021.114114
16. Tombácz D, Dörmő Á, Gulyás G, Csabai Z, Prazsák I, Kakuk B. High temporal resolution Nanopore sequencing dataset of SARS-CoV-2 and host cell RNAs. GigaScience. 2022;11:giac094. https://doi.org/10.1093/gigascience/giac094
17. Vacca D, Fiannaca A, Tramuto F, Cancila V, La Paglia L, Mazzucco W. Direct RNA Nanopore Sequencing of SARS-CoV-2 Extracted from Critical Material from Swabs. Life. 2022;12(1):69. https://doi.org/10.3390/life12010069
18. Gauthier NPG, Nelson C, Bonsall MB, Locher K, Charles M, MacDonald C. Nanopore metagenomic sequencing for detection and characterization of SARS-CoV-2 in clinical samples. Plos one. 2021;16(11):e0259712. https://doi.org/10.1371/journal.pone.0259712
19. Ben Chehida S, Filloux D, Fernandez E, Moubset O, Hoareau M, Julian C. Nanopore Sequencing Is a Credible Alternative to Recover Complete Genomes of Geminiviruses. Microorganisms. 2021;9(5):903. https://doi.org/10.3390/microorganisms9050903
20. Zhang, JY, Zhang, Y, Wang L, Guo F, Yun Q, Zeng T, Dong Y. A single-molecule nanopore sequencing platform. bioRxiv. 2024;08. https://doi.org/10.1101/2024.08.19.608720
21. Linde J, Brangsch H, Hölzer M, Thomas C, Elschner MC, Melzer F, et al. Comparison of Illumina and Oxford Nanopore Technology for genome analysis of Francisella tularensis, Bacillus anthracis, and Brucella suis. BMC Genomics.2023;24(1):258. https://doi.org/10.1186/s12864-023-09343-z
22. Satam H, Joshi K, Mangrolia U, Waghoo S, Zaidi G, Rawool S, et al. Next-Generation Sequencing Technology: Current Trends and Advancements. Biology. 2023;12(7):997. https://doi.org/10.3390/biology12070997
23. Greig DR, Jenkins C, Gharbia S, Dallman TJ. Comparison of single-nucleotide variants identified by Illumina and Oxford Nanopore technologies in the context of a potential outbreak of Shiga toxin–producing Escherichia coli. GigaScience. 2019;8(8):giz104. https://doi.org/10.1093/gigascience/giz104
24. Winand R, Bogaerts B, Hoffman S, Lefevre L, Delvoye M, Van Braekel J, et al.Targeting the 16S rRNA Gene for Bacterial Identification in Complex Mixed Samples: Comparative Evaluation of Second (Illumina) and Third (Oxford Nanopore Technologies) Generation Sequencing Technologies. Int J Mol Sci. 2020;21(1):298. https://doi.org/10.3390/ijms21010298
25. de Souza LM, de Oliveira ID, Sales FCS, da Costa AC, Campos KR, Abbud A, et al. Technical comparison of MinIon and Illumina technologies for genotyping Chikungunya virus in clinical samples. J Genet Eng Biotechnol. 2023;21:88. https://doi.org/10.1186/s43141-023-00536-3
26. Ye F, Han Y, Zhu J, Li P, Zhang Q, Lin Y, и др. First Identification of Human Adenovirus Subtype 21a in China With MinION and Illumina Sequencers. Front Genet. 2020. https://doi.org/10.3389/fgene.2020.00285
27. Peng K, Yin Y, Li Y, Qin S, Liu Y, Yang X, et al. QitanTech Nanopore Long-Read Sequencing Enables Rapid Resolution of Complete Genomes of Multi-Drug Resistant Pathogens. Frontiers in microbiology. 2022;13:778659. https://doi.org/10.3389/fmicb.2022.778659
28. Wang Z, Qin L, Liu J, Jiang L, Zou X, Chen X, et al. Forensic nanopore sequencing of microhaplotype markers using QitanTech’s QNome. Forensic science international. Genetics. 2022;57:102657. https://doi.org/10.1016/j.fsigen.2021.102657
Diana A. Grigoryan
Moscow
Ivan F. Stetsenko
Moscow
Boris S. Gukov
Moscow
Alina D. Matsvay
Moscow
German A. Shipulin
Moscow
Grigoryan D.A., Stetsenko I.F., Gukov B.S., Matsvay A.D., Shipulin G.A. Comparative evaluation of MinION and Nanoporus nanopore sequencers in identification of pathogen nucleic acids. Extreme Medicine. 2025;27(1):64-73. https://doi.org/10.47183/mes.2024-242
10 bld. 1 Pogodinskaya Str., Moscow, Russia 119121
tel.: +7 (495) 540-61-71, ext.: 4190, 4191, 4192
E-mail: Extrememedicine@cspfmba.ru
Processing of personal data












